Articles

Article Tools
 View Full Text View Full Text |
 Abstract Abstract |
 Article as PDF Article as PDF |
 Print this Article Print this Article |
 Pubmed Pubmed |
 PMC PMC |
 PubReader PubReader |
 Export to Citation Export to Citation |
 Email Alerts Email Alerts |
 Open Access Open Access |
Share this article on :



Stats or Metrics
Article
Review Article
Exp Neurobiol 2015; 24(4): 325-340
Published online December 30, 2015
https://doi.org/10.5607/en.2015.24.4.325
© The Korean Society for Brain and Neural Sciences
The Role of Oxidative Stress in Neurodegenerative Diseases
Geon Ha Kim1,2, Jieun E. Kim1,3, Sandy Jeong Rhie1,4 and Sujung Yoon1*
1Ewha Brain Institute, Ewha Womans University,
2Department of Neurology, Ewha Womans University Mokdong Hospital, Ewha Womans University School of Medicine,
3Department of Brain and Cognitive Sciences,
4College of Pharmacy, Graduate School of Pharmaceutical Sciences, Ewha Womans University, Seoul 03760, Korea
Correspondence to: *To whom correspondence should be addressed.
TEL: 82-2-3277-2466, FAX: 82-2-3277-6562
e-mail: sujungjyoon@ewha.ac.kr
Abstract
Oxidative stress is induced by an imbalanced redox states, involving either excessive generation of reactive oxygen species (ROS) or dysfunction of the antioxidant system. The brain is one of organs especially vulnerable to the effects of ROS because of its high oxygen demand and its abundance of peroxidation-susceptible lipid cells. Previous studies have demonstrated that oxidative stress plays a central role in a common pathophysiology of neurodegenerative diseases such as Alzheimer's disease and Parkinson's disease. Antioxidant therapy has been suggested for the prevention and treatment of neurodegenerative diseases, although the results with regard to their efficacy of treating neurodegenerative disease have been inconsistent. In this review, we will discuss the role of oxidative stress in the pathophysiology of neurodegenerative diseases and
Keywords: Oxidative stress, Reactive oxygen species, Neurodegenerative disease, Alzheimer’s disease, Parkinson’s disease, Antioxidant
INTRODUCTION
Neurodegenerative diseases, as a heterogeneous group of disorders, are characterized by slowly progressive losses of neurons [1,2]. The etiology of neurodegenerative diseases has not yet been fully elucidated, however increased oxidative stress has been suggested as one of the potential common etiology in various neurodegenerative diseases. Cumulative oxidative stress may induce cellular damage, impairment of the DNA repair system [3], and mitochondrial dysfunction, all of which have been known as key factors in acceleration of aging process and the development of neurodegenerative disorders [2,4,5]. For these reasons, there have been continuing efforts to find the agents that can protect against oxidative damage and potentially treat neurodegenerative diseases. In this review, we focus to discuss the fundamental pathophysiological pathway of oxidative stress to the development of neurodegenerative diseases, especially in Alzheimer's disease (AD) and Parkinson's disease (PD). In addition, we will outline the present knowledge of available evidence in the prevention and treatment of neurodegenerative diseases and future directions for the potential of antioxidant supplementation with enhanced efficacy.
CHARACTERISTICS OF REACTIVE OXYGEN SPECIES
Types of reactive oxygen species (ROS)
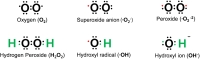
Oxygen is susceptible to radical formation due to two unpaired electrons present in the outer electron shell [6]. Reactive oxygen species (ROS) are defined as a group of reactive molecules derived from oxygen [7], which are generally short-lived and highly reactive because of their unpaired valence electrons [5]. ROS include, but are not limited to free radicals (superoxide, O2-), hydroxyl radical (·OH), or non-radicals (hydrogen peroxide, H2O2) (Fig. 1) [2,7,8].
O2- is suggested to play a gateway role in ROS production. O2- may be transformed into the more stable form of H2O2 by superoxide dismutases (SOD) (Fig. 2). It may also be protonated to form HO2-. H2O2 may have potential to generate highly reactive hydroxyl radicals ·OH [5], while it can further be divided into H2O and O2 by catalase, glutathione peroxidase, and other peroxidases [3] (Fig. 2). ·OH is known to be one of the most reactive ROS that are mainly responsible for the cytotoxic effects of ROS [7] ·OH can be generated from H2O2 and O2- and is catalyzed by iron ions through the Fenton reaction that refers to Fe2+-mediated decomposition of H2O2 [9].
Mitochondrial ROS production
The mitochondrion is the primary source of ROS production in the majority of cells. Under normal physiological condition, up to 2% of the total cellular mitochondrial O2 consumption may be related to the generation of ROS including O2- [11,12,13]. Multiple ways of mitochondrial ROS productions have been proposed, which are mainly modulated by the mitochondrial respiratory chain complexes [12,13,14]. The mitochondrial electron transport chain (ETC) consists of five multi-subunit complexes including NADH-coenzyme Q (CoQ) reductase (NADH dehydrogenase, Complex I), succinate dehydrogenase (Complex II), coenzyme Q-cytochrome c reductase (Complex III), cytochrome C oxidase (Complex IV), and ATP synthase (Complex V) [3,10].
Complex I is responsible for ROS production of O2- [7,15] and facilitates electron transfers from NADH to CoQ. During this step, protons are also translocated from the matrix to the intermembrane space [7]. Complex II is involved in the reduction of CoQ and is known to be involved in producing low levels of O2- [16,17]. Complex III, on the other hand, is involved in the generation of O2- in the intermembrane space. The generation of O2- is especially enhanced when the electron transfer is reduced with the increased membrane potential [7].
Interestingly, the capacity of these enzymes to produce ROS may vary among the organs or during disease conditions [18]. For instance, Complex I appears to contribute to the production of most of O2- in the brain, while Complex III is considered as the primary source of O2- in the heart and lung [18]. In addition, within mitochondria, ETC Complex I and III are regarded as the main producers of O2- [3]. ROS productions from Complex I is approximately one-half of those from complex III in heathy state [3], while Complex I exerts the primary role in ROS productions under pathological conditions ranging from accelerated aging to neurodegenerative diseases [9].
NADPH oxidases (Nox)
Nox, a transmembrane enzyme complex, is known to be another important endogenous source of O2- production as the result of the catalyzing the electron transfer from NADPH to oxygen [19,20]. Nox is found highly in phagocytes (neutrophils, eosinophils, monocytes and macrophages, called as Phox or NOX2) as well as in the endothelium of cardiovascular tissue [20,21]. Until now, seven Nox isoforms have been identified in mammalian cells including Nox1 to Nox5 and dual oxidases (Duox1 and Duox2) [21]. Each Nox isoform has unique cellular localization, regulation, and function [3,22]. For instance, Nox4 and Nox2 are abundant, whereas Nox1 is less in endothelial cells [23,24]. In contrast, Nox1 and Nox4 are the more highly expressed isoforms in vascular smooth muscle cell than Nox2 [25]. Nox2, which is mainly expressed in phagocytes and produces large amounts of ROS, can help kill the foreign organisms as a part of the immune defense system [5,20]. On the other hand, Nox produces relatively less ROS at a slow and sustained rate in cardiovascular tissue and exerts a role as intracellular signaling molecules [5]. Previous studies have reported that Nox4 is one of the most common isoforms in vascular structures [22,26,27]. In addition, contrary to Nox1 and Nox2, Nox4 is fundamentally active in the cardiovascular systems [28,29] and the primary source of H2O2 production rather than O2- production [30,31].
Xanthine oxidases (XO)
XO and xanthine dehydrogenase (XDH) are inter-convertible forms of xanthine oxidoreductase [32]. XO is responsible for the catabolism of purines by converting hypoxanthine to xanthine and xanthine to uric acid [33]. XO donates electrons to oxygen and subsequently generate O2- and H2O2 within the cell [34]. In normal conditions, the enzyme is present as a form of XDH [2]. The involvement of XO in ROS-mediated diseases has been proposed with the increased level of intracellular calcium when energy status of the cell decreases and transmembrane gradients are disrupted in such a case of ischemic injury. Increased intracellular calcium may lead to the irreversible conversion of XDH into XO that catalyzes the oxidation of hypoxanthine to xanthine [2,35]. Particularly during reperfusion, oxygen is reduced to its radical forms, H2O2 and O2-, in the presence of XO [2,34,35]. Increased ROS levels may contribute to further tissue damage.
ROS production in the endoplasmic reticulum
The endoplasmic reticulum (ER) is the membrane-based intracellular organelle that is primarily related to protein folding and lipid biosynthesis [10,36]. ER may generate ROS by following two mechanisms [36]. First, under normal conditions, proper formations of disulfide bonds and protein folding take place in the ER for the stability and maturation of the membrane, which is driven by endoplasmic reticulum oxidoreduction-1 (ERO-1) and protein disulfide-isomerase (PDI) [36]. As electrons are transferred from protein thiol to oxygen by ERO-1 and PDI [36], ROS can be produced as a byproduct. Second, ROS is also produced by protein misfolding particularly in cases of glutathione (GSH) depletion [36]. Oxidized thiols are repaired to interact with ERO-1 and PDI by glutathione (GSH) [36]. These steps would initiate repetitive cycles of breakage and formation of disulfide bond within ER lumen, which more ROS is generating as a byproduct [36,37]. Therefore, proteins which have multiple disulfide bonds may be more vulnerable to producing ROS [36]. Since GSH is used to reduce incorrectly formed disulfide bonds particularly under oxidizing environment, the level of GSH is further decreased and more ROS can be generated in this situation [30]. Furthermore, because the process involved in oxidative protein folding in the ER may be highly energy dependent, it should be noted that depletion of adenosine triphosphate (ATP) caused by protein misfolding may elicit oxidative phosphorylation in mitochondria and consequently produce more ROS [36,38].
ROS production in peroxisomes
Peroxisomes are present in most eukaryotic cells and participate in multiple metabolic pathways including fatty acid oxidation, phospholipid biosynthesis, amino acid catabolism, and oxidative part of the pentose phosphate pathway [10,39]. Under normal physiologic conditions, oxygen consumption in the peroxisomes may lead to the production of H2O2, but not O2-. Peroxisomes is one of the organelles that can generate the majority of H2O2 using a number of different oxidases such as acyl-CoA oxidases, D-aspartate oxidase, and urate oxidase, which makes hydrogen to be transferred to O2 from their respective substrates [10]. In addition, peroxisomes also contain catalases that can decompose H2O2 and help maintain the balance between the production and removal of ROS [39,40]. When peroxisomes are damaged and the amount of catalases decreases, increased H2O2 release into the cytosol can contribute to oxidative stress in the cell [40].
Superoxide dismutase (SOD)
SOD plays a significant role in catalyzing the breakdown of highly reactive O2- to less reactive H2O2 and oxygen [33]. Cytosolic copper/zinc-SOD (SOD1), mitochondrial manganese SOD (SOD2), and extracellular SOD (SOD3) are three distinct isoforms of SOD that have been identified. SOD1 and SOD2 are mainly involved in the elimination of O2- in the cytosol and mitochondria, respectively [33].
Glutathione peroxidases (GPX)
GPX contains a family of multiple isoenzymes which catalyze the reduction of H2O2 and lipid peroxides utilizing GSH as an electron donor [2,33]. GPX is located in both cytosol and mitochondria. In mammals, there are five different isoforms of selenium-dependent glutathione peroxidases (GPX1-4 and 6) and three non-selenium congeners (GPX 5, 7 and 8) that have cysteine instead of selenocysteine [42]. Antioxidant function of GPXs depends on each isoform and location in the cells; GPX1 exists universally in the cytosol and mitochondria, GPX2 does in the epithelium of intestine, and GPX3 does in the plasma [42]. It is noteworthy that GPX1 has been regarded as one of the major antioxidant enzymes in the brain, which is expressed predominantly in microglia but not in neurons [41]. Studies have suggested that upregulation of GPX1 could be one of the protective responses against neuronal injury [41].
Catalase
Catalase is responsible for the conversion of H2O2 to water and oxygen using either iron or manganese as a cofactor [2,33,43]. Catalase is located in peroxisomes and also found in the cytoplasm and mitochondria [2]. The role of catalase is minor at low levels of H2O2, but becomes increasingly important at higher levels of H2O2 [2].
Peroxiredoxins (PRX)
PRX are thiol-specific peroxidases that catalyze the reduction of H2O2 as well as other organic hydroperoxides and peroxynitrite [33,44,45]. Among the six PRX isoforms, PRX1, 2, and 4 are present in the cytoplasm as well as in the nuclei. In addition, PRX1 is also expressed in the mitochondria and peroxisomes, while PRX4 is found in the lysosomes [44]. PRX3 is exclusively localized in the mitochondria [44,46], whereas PRX5 is found in the mitochondria, cytoplasm, nuclei, and peroxisomes [47]. All PRX utilize a conserved active-site cysteine residue in order to directly reduce peroxide [48]. Since PRX are abundant in eukaryotic cells, constitute approximately more than 1% of cellular proteins, and show high reactivity, PRX are responsible for the reduction of up to 90% of mitochondrial H2O2 and almost 100% of cytoplasmic H2O2 [48,49,50,51].
Glutathione (GSH)
GSH, a tripeptide synthesized from glutamate, cysteine, and glycine, exerts protective function of cell survival against oxidative stress [2,33]. In the brain,
GSH is involved in the following two types of reactions; Firstly, GSH, in its reduced form, is known to non-enzymatically react with ROS such as O2- and ·OH for the removal of ROS [2,53]. Secondly, GSH is the electron donor for the reduction of peroxides in the GPX reaction [54]. Reaction with ROS firstly oxidizes GSH, which generates glutathione disulfide, the final product of GPX reactions. GSH can be regenerated from glutathione disulfide by the reaction with glutathione reductase that transfers electrons from NADPH to glutathione disulfide [54,55].
Several studies have reported that GSH is involved in inhibiting apoptotic cell death [32,56] and DNA damage in cells following oxidative stress [56,57].
Vitamin E
Vitamin E is a lipid-soluble antioxidant that can attenuate the effects of peroxide and protect against lipid peroxidation in cell membranes [2,33].
Vitamin C
Vitamin C is a water-soluble antioxidant, which is involved in the removal of free radicals by electron transfer and also acts as a cofactor for antioxidant enzymes [3,33].
Physiological functions of ROS
Low to moderate levels of ROS are critical in cellular signaling and pro-survival pathways [3,5,40,58]. For instance, Nox-derived ROS play a role in cellular signaling related to the cardiovascular systems [22] and those in phagocytes (Nox2-derived) are involved in defense mechanisms of the immune system against foreign organisms [20]. Furthermore, the increased level of Nox-derived ROS activates important survival pathways, such as mitogen-activated protein kinase (MAPK) pathways [5]. The MAPK, the serine/threonine-specific protein kinases, represents the major redox-regulated signaling molecules in the cardiovascular systems [59]. It also modulates various cellular activities including gene expression, mitosis, proliferation, migration, cell survival, and apoptosis [5,58,60].
ROS can also activate transcription factors that regulate cellular responses to ROS [5]. Increased ROS may therefore promote antioxidant defense processes. An example is NF-E2-related factor 2 (Nrf2), which is one of major redox-sensitive transcription factors. It is activated by ROS and modulates the expression of several antioxidant enzymes including SOD, PRX, GPX, and heme oxygenases [61,62]. A suppressor protein, Kelch-like ECH-associated protein 1 (Keap1), which is anchored in the cytoplasm, prevents the translocation of Nrf2 to the nucleus and keeps Nrf2 inactive under normal conditions [62]. Increased ROS production disrupts binding between Keap1 and Nrf2, allowing transcription by activation of Nrf2 [5,63]. Nuclear factor-kappa B (NF-κB) would be another pro-survival transcription factor that may be activated by ROS [5]. NF-κB is normally present in the cytoplasm as an inactive state by the action of a NF-κB inhibitor. Moderate levels of ROS may induce the phosphorylation and degradation of a NF-κB inhibitor and result in activation of NF-κB [64]. The activated NF-κB transcripts anti-apoptotic proteins and inhibits caspase-dependent cell death pathways [5,65]. In contrast, high levels of ROS may contribute to inactivation of NF-κB by inhibiting its binding to DNA, attenuate pro-survival pathway, and consequently promote apoptosis [66]. In this regard, the role of NF-κB activation in a survival response to apoptosis depends on the amount of ROS formation [65].
Oxidative stress: excessive accumulation of ROS
In a healthy condition, the production of ROS is balanced by various antioxidant systems [2,33]. Oxidative stress is a condition of imbalance between ROS production and antioxidant defenses, resulting in excessive accumulation of ROS [33,67]. Oxidative stress may be related to cell membrane damage from lipid peroxidation, changes in protein structure and function due to protein oxidation, and structural damage to DNA [2].
As the brain is one of the most metabolically active organs in the body, it is vulnerable to oxidative stress particularly because of the following reasons. First, the brain has a high oxygen demand, which constitutes 20% of the body oxygen consumption. Second, the redox-active metals such as iron or copper exist abundantly in the brain and they are actively involved to catalyze ROS formation. Third, the high levels of polyunsaturated fatty acids are found in the brain cell membranes and react as substrates for lipid peroxidation [68]. Fourth, there are relatively low levels of GSH in the brain, which plays a role of endogenous antioxidant in the elimination of ROS [69].
OXIDATIVE STRESS AND NEURODEGENERATIVE DISEASES
Alzheimer's disease
Alzheimer's disease (AD), as one of the most common neurodegenerative diseases, is characterized by progressive neuronal loss and accumulation of proteins including extracellular amyloid plaques (Aβ) and intracellular tau tangles (neurofibrillary tangles, NFT) [70]. It has been suggested that oxidative imbalance and resultant neuronal damage may play a critical role in the initiation and progression of AD [71]. The excessive accumulation of ROS in patients with AD may induce mitochondrial dysfunction, however, the origin of increased ROS production and the exact mechanisms underlying the disruption of redox balance still remain elusive [72].
The accumulation of Aβ seems to increase oxidative stress and lead to mitochondrial dysfunction and energy failure [2] even in early stage of AD [73]. Previous studies have implicated that Aβ-induced oxidative imbalance may increase the levels of the byproducts related to lipid peroxidation (e.g. 4-hydroxynonal, malonidialdehyde), protein oxidation (e.g. carbonyl) and DNA/RNA oxidation (e.g. 8-hydroxyldeoxyguanosine and 8-hydroxylguanosine). In contrast, decreased levels of antioxidants (e.g. uric acid, vitamin C and E) or antioxidant enzymes (e.g. superoxide dismutase, catalase etc.) have been found in patients with AD [71,72]. In addition, AD transgenic mouse models expressing mutant amyloid precursor protein (APP) and presenilin-1 (PS-1) have shown increased levels of H2O2 and peroxidation of proteins and lipids, implying that Aβ may enhance oxidative stress in AD [72,74].
Oxidative stress can also aggravate the production and aggregation of Aβ and promote the phosphorylation of tau protein, which could induce a vicious cycle of pathogenesis in AD [72]. Numerous previous studies have proved that oxidative stress also promotes the production of Aβ. It was reported that the defects in antioxidant defense mechanisms caused increased oxidative stress and, further facilitated Aβ depositions in transgenic mice with APP mutation [72]. Moreover, the deletion of cytoplasmic/zinc SOD in the Tg2576 APP overexpressing AD mouse model was related to increases in the oligomerization of Aβ and aggravated memory dysfunction [72,75]. It has been hypothesized that oxidative stress may decrease the activity of α-secretase, promote the expression and activity of β and γ-secretase, and then lead to the enhanced production of Aβ [76].
There is emerging evidence to suggest the relationship between oxidative stress and tau pathology. It has been reported that cells with overexpressed tau protein showed increased vulnerability to oxidative stress, and it may be due to the depletion of peroxisomes [72,77]. Furthermore, transgenic mouse models expressing mutant (P301S and P301L) tau proteins showed reduced NADH-ubiquinone oxidoreductase activity and mitochondrial dysfunction, both of which were associated with increased ROS production [78,79,80].
Aβ is accumulated primarily in the extracellular regions but also found in different subcellular areas including the ER and Golgi apparatus [81]. Interestingly, previous studies on AD have shown that the accumulation of Aβ has also been observed in the mitochondria [81], which may affect mitochondrial respiratory function, increase ROS production, and change mitochondrial membrane potentials in various brain regions [81]. Aβ-induced mitochondrial dysfunction has been suggested to inhibit the efficient production of ATP and increase the generation of ROS in AD [72,82].
There are several evidences that suggest mitochondrial dysfunction in AD. First, reduced energy metabolism of the brain has frequently been observed in AD [71]. A previous study has also reported that decreased cerebral glucose metabolism in AD was associated with reductions in neuronal expression of genes that encode subunits of the mitochondrial electron transport chain [71]. Second, the activities of key enzymes of oxidative metabolism including α-ketoglutarate dehydrogenase complex, pyruvate dehydrogenase complex, and cytochrome oxidase were reduced in patients with AD [71]. They were significantly correlated with the clinical severity and the senile plaque [83]. Reduced Complex IV activity has also been observed in the mitochondria of the hippocampus and platelets in patients with AD [2]. Third, Aβ-induced mitochondrial dysfunction contributes to impairment in calcium homeostasis [71]. This process is observed subsequently with the result in increased calcium overload and decreased reuptake of calcium [84]. The accumulation of mitochondrial calcium may be related to increased ROS production and the opening of permeability transition pore (PTP) [2]. It may be responsible for the translocation of pro-apoptotic molecules from the mitochondria to the cytosol and apoptosis. Increases in intracellular calcium can be measured indirectly by assessing the activity of calmodulin-dependent kinase and calpain. Interestingly, increased activity in calmodulin-dependent kinase and calpain has been observed in the early stage of AD [71]. Finally, increased oxidative damage to mitochondrial DNA, which may lead to more mutations of mitochondrial DNA, has been reported in patients with AD [71].
Parkinson's disease
Parkinson's disease (PD) is a neurodegenerative disorder characterized by selective neuronal loss of dopaminergic (DA) neurons in the substantia nigra pars compacta (SNc) and decreased DA levels in the nigrostriatal DA pathway in the brain [85,86]. Although the exact mechanism still remains unclear, oxidative stress has been considered as one of major pathophysiological mechanisms underlying PD [86,87].
Previous studies have found the reduced activity in Complex I of the respiratory chain in SNc of patients with PD, which may contribute to the generation of excessive ROS and, in turn, induce apoptosis [86,87,88]. Mitochondrial Complex I deficiency in the frontal cortex, fibroblasts, and blood platelets have also been reported in patients with PD [86]. Furthermore, the relationship between mitochondrial dysfunction and PD is supported by the findings that the Complex I inhibitors, such as 1-methyl-4-phenyl-1,2,3,4-tetrahydropyridine (MPTP) and its metabolite, 1-methyl-4-phenylpyridinium (MPP+), may exert cytotoxic effects on the DA neurons, resultong in clinically parkinsonian phenotype, and induce nigral degeneration with cytoplasmic α-synuclein [1]. It has also been reported that genetic mutations in proteins including α-synuclein, parkin, and phosphatase and tensin homolog-induced putative kinase (PINK) were linked to the familial forms of PD [86]. Mutations of these genes have been known to affect mitochondrial function and increase oxidative stress [86].
Increasing evidence has supported that the accumulation of point mutations and deletions in mitochondrial DNA may be associated with Complex I defect or mitochondrial dysfunction in PD [89]. Mitochondrial DNA has been well known to encode 13 proteins, mitochondrial transfer RNA, and ribosomal RNA including subunits from electron transport chain. Point mutations or deletions in mitochondrial DNA encoding Complex I subunit have been reported in patients with PD [89,90,91].
Changes in the antioxidant molecules have also been reported even in the early stage of PD [89]. For example, the levels of GSH, a major antioxidant molecule, have been reduced in the SNc of PD although this finding is not specific for PD [92]. In addition, higher levels of iron were observed in the SNc of PD in comparison with that of controls, which may arise from dysfunctional transport of iron to the mitochondria in DA neurons of PD [93,94] . It has also been reported that iron levels were increased in DA neurons in PD, which may allow easier interaction of ferrous iron with H2O2 and enhance production of highly toxic hydroxyl radicals (OH) [89]. High levels of iron in the SNc, therefore, may potentially exert harmful effects on the survivals of DA neurons
EVALUATION OF OXIDATIVE STRESS IN NEURODEGENERATIVE DISEASES
Measurement of oxidative stress in peripheral blood
Since oxidative stress may be a common pathophysiological mechanism underlying various neurodegenerative diseases, several surrogate markers for oxidative stress or antioxidant activity, including circulating lipid peroxides, GSH, and vitamins C and E, have been assessed in peripheral blood [69,95,96,97,98]. A previous study has shown that AD patients demonstrated the decreased peripheral levels of vitamins A, C, and E [97] along with lower activities of SOD and glutathione peroxidase [95]. The levels of GSH in plasma have been suggested as a significant predictor of cognitive functions in patients with AD, implying the relationship between lower plasma levels of GSH and more severe cognitive impairment [99]. Although the results have been inconsistent, the activity of SOD in erythrocyte would be altered in patients in PD [100,101,102,103]. Increased SOD activity may contribute to the protection mechanism against enhanced production of O2- relating to neurodegenerative diseases [104].
Magnetic Resonance Spectroscopy
Glutathione (GSH)
GSH is currently the only antioxidant which can be measured using 1H magnetic resonance spectroscopy (MRS) [105]. Until recently, the assessment of GSH in the human brain has been quite challenging since the concentration of GSH is lower in the human brain than those of other metabolites including N-acetyl aspartate, creatine, and choline. In addition, it is difficult to distinguish between the spectral proximity of resonance peak in GSH and those in other metabolites such as glutamate with MRS [105,106]. For example, two protons from β-CH2 of GSH-cysteine resonate at 2.93 and 2.97 ppm, which largely overlap with those of creatine (3.03 ppm) and aspartate (2.82 ppm) [105]. Therefore, in order to enhance GSH signal and to acquire reliable signals from the nuclei, specific spectral editing techniques such as MEscher-GArwood-PRESS (MEGA-PRESS) [107] would be required to measure GSH levels in the brain. The MEGA-PRESS technique in combination with additional editing pulse of 180o in the original PRESS pulse sequence [108] can help separate GSH-cysteine signals from other signals, especially the creatine signals in the brain [109]. Using this technique, a recent study has reported that the concentration of GSH was reduced in patients with AD, particularly in hippocampus and frontal cortices [108,110]. These reductions were correlated with global cognitive functions [108,110].
Vitamin C (Ascorbic acid)
The concentration of vitamin C is approximately 1.0 mM in the human brain, which can be detectable with the use of MRS [105]. However, it is also difficult to measure vitamin C with 1H MRS due to the similarity between the resonance of vitamin C (3.73, 4.01 and 4.50 ppm) and that of glutamate (3.75 ppm) [105]. The MEGA-PRESS editing could also help measure vitamin C in the human brain [111]. A few previous studies using 1H-MRS with the MEGA-PRESS have measured the levels of vitamin C in the human brain [105,109,111].
Positron Emission Tomography
[62Cu] diacetyl-bis (N4-methylthiosemicarbazone) ([62Cu] ATSM)
[62Cu] diacetyl-bis (N4-methylthiosemicarbazone) ([62Cu] ATSM) is a radiotracer for positron emission tomography (PET) that can measure intracellular over-reductive state [112]. This PET tracer has previously been used to detect an over-reductive state from myocardial ischemia or hypoxia associated with malignant tumors [113]. [62Cu] ATSM could be accumulated in the brain regions in an over-reductive state through the reduction of Cu (II) to CU (I) [114,115]. Recently, regional oxidative stress which was mainly caused by mitochondrial dysfunction in patients with PD has been visualized through the application of this tracer in one study. In this study, the deposition of [62Cu] ATSM was observed in the striatum in patients with PD [113], suggesting regional over-reductive state induced by mitochondrial dysfunction.
Electron paramagnetic resonance (EPR) spectroscopy
EPR spectroscopy has been considered as one of possible methods to detect and characterize the radicals
ANTIOXIDANT TREATMENT FOR NEURODEGENERATIVE DISEASES
Clinical studies with antioxidant therapy in neurodegenerative diseases
Antioxidants are divided into endogenous or exogenous agents. The human endogenous antioxidant molecules include various enzymes such as SOD, GPX and catalase as well as non-enzymatic molecules (e.g. uric acid, GSH, and ascorbic acid), precursors of antioxidants (e.g. N-acetyl cystein) and cofactors of antioxidants (e.g. selenium and coenzyme Q 10) [119]. The exogenous antioxidants could be natural (e.g. N-acetyl cysteine, NAC) or synthetic (e.g. α-Lipoic Acid) [120]. The most commonly used antioxidants for clinical applications include vitamin E (the important scavenger of lipid peroxidation in the brain), vitamin C (intracellular reducing molecule), NAC (acting as a precursor of GSH), and coenzyme Q10 (transporter of electrons from complexes I and II to III in the ETC).
Although the initial results of the efficacy of antioxidants in the animal studies have been promising, the majority of the clinical trials in humans have shown negative results in terms of their efficacy for neurodegenerative diseases. A summary of clinical studies for neurodegenerative diseases is presented in Table 1 [121,122,123,124,125,126,127,128,129,130,131,132,133,134,135,136,137].
Possible reasons for little efficacy of antioxidants in treating neurodegenerative diseases
The following explanations may address why the current clinical trials have not yet found the potential antioxidants, which would effectively treat neurodegenerative diseasess.
First, antioxidant therapy could not decrease oxidative stress in patients with neurodegenerative diseases potentially due to insufficient dose of antioxidants, unsuitable timing for therapy, or inappropriate duration of treatments [138]. On each related issue, the actual challenge could be how to evaluate the exact effects of antioxidants on the levels of a particular ROS at its proper action sites [138]. It is also important to evaluate the magnitude of therapeutic effects of antioxidants on alterations in levels of a particular ROS at its presumptively proper action sites [138]. In that sense, the development of biomarkers to better assess ROS is critical for the development of novel antioxidant therapeutic approach of neurodegenerative diseases.
The second point is that the oxidative damage may not be the primary cause, which contributes to the pathophysiology of the neurodegenerative diseases. An increase in oxidative damage could occur during the progression of disease, but it does not necessarily mean that it certainly causes the disease [138]. If so, antioxidants would not be the appropriate target for the treatment of neurodegenerative diseases. In addition, oxidative stress may not be the only cause of the disease. It should be noted that other deleterious processes such as inflammation and excitotoxicity also could be involved in the pathogenesis of neurodegenerative disease [139]. Therefore, it would be necessary to apply the combination of antioxidants with other drugs or multifunctional agents in treating neurodegenerative diseases.
Third, it is plausible that one single antioxidant may not be sufficient to resist the oxidative damage since the oxidative stress is modulated by a complex system of endogenous and exogenous antioxidants. In this regard, the combinatory approach of antioxidants would be necessary to be studied in the treatment of neurodegenerative diseases [120,138].
Fourth, the timing of antioxidant therapy might not be optimal in previous clinical trials. Most trials have assessed the efficacy of antioxidants in patients with advanced AD or PD. Given that antioxidants may exert a prophylactic role in neurodegenerative disease, the earlier application of antioxidants, even before the onset of symptoms, may be effective [120,139]. The optimization of time and duration of antioxidant therapy would be necessary to be elucidated.
Fifth, inter-individual differences in the levels of endogenous antioxidants may affect responses to antioxidant therapy [139]. Before starting antioxidant treatment, it might be helpful to select participants who could be the potential responders such as those with low levels of endogenous antioxidants, rather than the probable non-responders with high or normal levels of endogenous antioxidants [140].
CONCLUSIONS
The role of oxidative stress in the pathogenesis of neurodegenerative diseases has been well demonstrated in many preclinical and clinical studies. However, the benefit of antioxidant therapy for neurodegenerative diseases is still controversial in human, although the pre-clinical studies have shown promising results. One of reasons for such discrepancy would be that there was no effective measurement of oxidative stress in the brain. Unfortunately, peripheral biomarkers may not necessarily represent oxidative stress in the brain and changes in neuronal function. Therefore, proper central biomarkers for oxidative stress should be identified to detect objective benefits to the brain and find exact therapeutic targets in treating neurodegenerative diseases.
Figures
{kind=link}

{kind=link}
Tables
{kind=link}
DPRCT: double-blind, placebo-controlled, randomized clinical trial, MAOI: monoamine oxidase inhibitor, ADCS-ADL: alzheimer's disease cooperative study-activities of daily living, MMSE: mini-mental status examination, ADAS-total: alzheimer's disease assessment scale-total score, ADAS-Cog: alzheimer's disease assessment scale-cognitive subcomponent, CGIC: clinical global impression of change, UPDRS: unified parkinson disease rating scale.
References
- Lin MT, Beal MF. Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases. Nature 2006;443:787-795.

- Gandhi S, Abramov AY. Mechanism of oxidative stress in neurodegeneration. Oxid Med Cell Longev 2012;2012:428010.
- Song P, Zou MH. In: Wang H, Patterson C. Atherosclerosis: risks, mechanisms, and therapies. Hoboken, NJ: John Wiley & Sons Inc., 2015; 2015. p. 379-392.
- Federico A, Cardaioli E, Da Pozzo P, Formichi P, Gallus GN, Radi E. Mitochondria, oxidative stress and neurodegeneration. J Neurol Sci 2012;322:254-262.
- Patten DA, Germain M, Kelly MA, Slack RS. Reactive oxygen species: stuck in the middle of neurodegeneration. J Alzheimers Dis 2010;20:S357-S367.
- Held P. An introduction to reactive oxygen species: measurement of ROS in cells (white paper). BioTek Instruments, Inc.: Winooski, VT, 2012.
- Bolisetty S, Jaimes EA. Mitochondria and reactive oxygen species: physiology and pathophysiology. Int J Mol Sci 2013;14:6306-6344.
- Halliwell B. Oxidative stress and neurodegeneration: where are we now?. J Neurochem 2006;97:1634-1658.
- Zorov DB, Juhaszova M, Sollott SJ. Mitochondrial reactive oxygen species (ROS) and ROS-induced ROS release. Physiol Rev 2014;94:909-950.
- Mani S. In: Rani V, Yadav UC. Free radicals in human health and disease. New Delhi: Springer, 2015; 2015. p. 3-15.
- Orrenius S. Reactive oxygen species in mitochondria-mediated cell death. Drug Metab Rev 2007;39:443-455.
- Widlansky ME, Gutterman DD. Regulation of endothelial function by mitochondrial reactive oxygen species. Antioxid Redox Signal 2011;15:1517-1530.
- Cadenas E, Davies KJ. Mitochondrial free radical generation, oxidative stress, and aging. Free Radic Biol Med 2000;29:222-230.
- Mancuso M, Coppede F, Migliore L, Siciliano G, Murri L. Mitochondrial dysfunction, oxidative stress and neurodegeneration. J Alzheimers Dis 2006;10:59-73.
- Quinlan CL, Perevoshchikova IV, Hey-Mogensen M, Orr AL, Brand MD. Sites of reactive oxygen species generation by mitochondria oxidizing different substrates. Redox Biol 2013;1:304-312.
- McLennan HR, Degli Esposti M. The contribution of mitochondrial respiratory complexes to the production of reactive oxygen species. J Bioenerg Biomembr 2000;32:153-162.
- Yankovskaya V, Horsefield R, Törnroth S, Luna-Chavez C, Miyoshi H, Léger C, Byrne B, Cecchini G, Iwata S. Architecture of succinate dehydrogenase and reactive oxygen species generation. Science 2003;299:700-704.
- Turrens JF. Mitochondrial formation of reactive oxygen species. J Physiol 2003;552:335-344.
- Infanger DW, Sharma RV, Davisson RL. NADPH oxidases of the brain: distribution, regulation, and function. Antioxid Redox Signal 2006;8:1583-1596.
- Babior BM. NADPH oxidase. Curr Opin Immunol 2004;16:42-47.
- Lambeth JD. NOX enzymes and the biology of reactive oxygen. Nat Rev Immunol 2004;4:181-189.
- Song P, Zou MH. Regulation of NAD(P)H oxidases by AMPK in cardiovascular systems. Free Radic Biol Med 2012;52:1607-1619.
- Görlach A, Brandes RP, Nguyen K, Amidi M, Dehghani F, Busse R. A gp91phox containing NADPH oxidase selectively expressed in endothelial cells is a major source of oxygen radical generation in the arterial wall. Circ Res 2000;87:26-32.
- Bengtsson SH, Gulluyan LM, Dusting GJ, Drummond GR. Novel isoforms of NADPH oxidase in vascular physiology and pathophysiology. Clin Exp Pharmacol Physiol 2003;30:849-854.
- Wingler K, Wünsch S, Kreutz R, Rothermund L, Paul M, Schmidt HH. Upregulation of the vascular NAD(P) H-oxidase isoforms Nox1 and Nox4 by the renin-angiotensin system in vitro and in vivo. Free Radic Biol Med 2001;31:1456-1464.
- Van Buul JD, Fernandez-Borja M, Anthony EC, Hordijk PL. Expression and localization of NOX2 and NOX4 in primary human endothelial cells. Antioxid Redox Signal 2005;7:308-317.
- Clempus RE, Sorescu D, Dikalova AE, Pounkova L, Jo P, Sorescu GP, Schmidt HH, Lassègue B, Griendling KK. Nox4 is required for maintenance of the differentiated vascular smooth muscle cell phenotype. Arterioscler Thromb Vasc Biol 2007;27:42-48.
- Ellmark SH, Dusting GJ, Fui MN, Guzzo-Pernell N, Drummond GR. The contribution of Nox4 to NADPH oxidase activity in mouse vascular smooth muscle. Cardiovasc Res 2005;65:495-504.
- Bedard K, Krause KH. The NOX family of ROS-generating NADPH oxidases: physiology and pathophysiology. Physiol Rev 2007;87:245-313.
- Gordillo G, Fang H, Park H, Roy S. Nox-4-dependent nuclear H2O2 drives DNA oxidation resulting in 8-OHdG as urinary biomarker and hemangioendothelioma formation. Antioxid Redox Signal 2010;12:933-943.
- Dikalov SI, Dikalova AE, Bikineyeva AT, Schmidt HH, Harrison DG, Griendling KK. Distinct roles of Nox1 and Nox4 in basal and angiotensin II-stimulated superoxide and hydrogen peroxide production. Free Radic Biol Med 2008;45:1340-1351.
- Abramov AY, Scorziello A, Duchen MR. Three distinct mechanisms generate oxygen free radicals in neurons and contribute to cell death during anoxia and reoxygenation. J Neurosci 2007;27:1129-1138.
- Dasuri K, Zhang L, Keller JN. Oxidative stress, neurodegeneration, and the balance of protein degradation and protein synthesis. Free Radic Biol Med 2013;62:170-185.
- Harrison R. Structure and function of xanthine oxidoreductase: where are we now?. Free Radic Biol Med 2002;33:774-797.
- Harrison R. Physiological roles of xanthine oxidoreductase. Drug Metab Rev 2004;36:363-375.
- Bhandary B, Marahatta A, Kim HR, Chae HJ. An involvement of oxidative stress in endoplasmic reticulum stress and its associated diseases. Int J Mol Sci 2012;14:434-456.
- Higa A, Chevet E. Redox signaling loops in the unfolded protein response. Cell Signal 2012;24:1548-1555.
- Malhotra JD, Kaufman RJ. Endoplasmic reticulum stress and oxidative stress: a vicious cycle or a double-edged sword?. Antioxid Redox Signal 2007;9:2277-2294.
- Schrader M, Fahimi HD. Peroxisomes and oxidative stress. Biochim Biophys Acta 2006;1763:1755-1766.
- Valko M, Leibfritz D, Moncol J, Cronin MT, Mazur M, Telser J. Free radicals and antioxidants in normal physiological functions and human disease. Int J Biochem Cell Biol 2007;39:44-84.
- Power JH, Blumbergs PC. Cellular glutathione peroxidase in human brain: cellular distribution, and its potential role in the degradation of Lewy bodies in Parkinson's disease and dementia with Lewy bodies. Acta Neuropathol 2009;117:63-73.
- Brigelius-Flohé R, Maiorino M. Glutathione peroxidases. Biochim Biophys Acta 2013;1830:3289-3303.
- Dröge W. Free radicals in the physiological control of cell function. Physiol Rev 2002;82:47-95.
- Espinosa-Diez C, Miguel V, Mennerich D, Kietzmann T, Sánchez-Pérez P, Cadenas S, Lamas S. Antioxidant responses and cellular adjustments to oxidative stress. Redox Biol 2015;6:183-197.
- Wood ZA, Schröder E, Robin Harris J, Poole LB. Structure, mechanism and regulation of peroxiredoxins. Trends Biochem Sci 2003;28:32-40.
- Oberley TD, Verwiebe E, Zhong W, Kang SW, Rhee SG. Localization of the thioredoxin system in normal rat kidney. Free Radic Biol Med 2001;30:412-424.
- Seo MS, Kang SW, Kim K, Baines IC, Lee TH, Rhee SG. Identification of a new type of mammalian peroxiredoxin that forms an intramolecular disulfide as a reaction intermediate. J Biol Chem 2000;275:20346-20354.
- Hall A, Nelson K, Poole LB, Karplus PA. Structure-based insights into the catalytic power and conformational dexterity of peroxiredoxins. Antioxid Redox Signal 2011;15:795-815.
- Winterbourn CC, Metodiewa D. The reaction of superoxide with reduced glutathione. Arch Biochem Biophys 1994;314:284-290.
- Winterbourn CC. Reconciling the chemistry and biology of reactive oxygen species. Nat Chem Biol 2008;4:278-286.
- Marinho HS, Real C, Cyrne L, Soares H, Antunes F. Hydrogen peroxide sensing, signaling and regulation of transcription factors. Redox Biol 2014;2:535-562.
- Lee M, Cho T, Jantaratnotai N, Wang YT, McGeer E, McGeer PL. Depletion of GSH in glial cells induces neuro-toxicity: relevance to aging and degenerative neurological diseases. FASEB J 2010;24:2533-2545.
- Dringen R, Gutterer JM, Hirrlinger J. Glutathione metabolism in brain metabolic interaction between astrocytes and neurons in the defense against reactive oxygen species. Eur J Biochem 2000;267:4912-4916.
- Dringen R. Metabolism and functions of glutathione in brain. Prog Neurobiol 2000;62:649-671.
- Dringen R, Hirrlinger J. Glutathione pathways in the brain. Biol Chem 2003;384:505-516.
- Presnell CE, Bhatti G, Numan LS, Lerche M, Alkhateeb SK, Ghalib M, Shammaa M, Kavdia M. Computational insights into the role of glutathione in oxidative stress. Curr Neurovasc Res 2013;10:185-194.
- Song J, Kang SM, Lee WT, Park KA, Lee KM, Lee JE. Glutathione protects brain endothelial cells from hydrogen peroxide-induced oxidative stress by increasing nrf2 expression. Exp Neurobiol 2014;23:93-103.
- Groeger G, Quiney C, Cotter TG. Hydrogen peroxide as a cell-survival signaling molecule. Antioxid Redox Signal 2009;11:2655-2671.
- Manea A. NADPH oxidase-derived reactive oxygen species: involvement in vascular physiology and pathology. Cell Tissue Res 2010;342:325-339.
- Lassègue B, Griendling KK. NADPH oxidases: functions and pathologies in the vasculature. Arterioscler Thromb Vasc Biol 2010;30:653-661.
- de Vries HE, Witte M, Hondius D, Rozemuller AJ, Drukarch B, Hoozemans J, van Horssen J. Nrf2-induced antioxidant protection: a promising target to counteract ROS-mediated damage in neurodegenerative disease?. Free Radic Biol Med 2008;45:1375-1383.
- Hybertson BM, Gao B, Bose SK, McCord JM. Oxidative stress in health and disease: the therapeutic potential of Nrf2 activation. Mol Aspects Med 2011;32:234-246.
- Itoh K, Wakabayashi N, Katoh Y, Ishii T, Igarashi K, Engel JD, Yamamoto M. Keap1 represses nuclear activation of antioxidant responsive elements by Nrf2 through binding to the amino-terminal Neh2 domain. Genes Dev 1999;13:76-86.
- Bubici C, Papa S, Dean K, Franzoso G. Mutual cross-talk between reactive oxygen species and nuclear factor-kappa B: molecular basis and biological significance. Oncogene 2006;25:6731-6748.
- Kriete A, Mayo KL. Atypical pathways of NF-kappaB activation and aging. Exp Gerontol 2009;44:250-255.
- Toledano MB, Leonard WJ. Modulation of transcription factor NF-kappa B binding activity by oxidation-reduction in vitro. Proc Natl Acad Sci U S A 1991;88:4328-4332.
- Ray PD, Huang BW, Tsuji Y. Reactive oxygen species (ROS) homeostasis and redox regulation in cellular signaling. Cell Signal 2012;24:981-990.
- Wang X, Michaelis EK. Selective neuronal vulnerability to oxidative stress in the brain. Front Aging Neurosci 2010;2:12.
- Ferreira ME, de Vasconcelos AS, da Costa Vilhena T, da Silva TL, da Silva Barbosa A, Gomes AR, Dolabela MF, Percário S. Oxidative Stress in Alzheimer's Disease: Should We Keep Trying Antioxidant Therapies?. Cell Mol Neurobiol 2015;35:595-614.
- Mattson MP. Pathways towards and away from Alzheimer's disease. Nature 2004;430:631-639.
- Wang X, Wang W, Li L, Perry G, Lee HG, Zhu X. Oxidative stress and mitochondrial dysfunction in Alzheimer's disease. Biochim Biophys Acta 2014;1842:1240-1247.
- Zhao Y, Zhao B. Oxidative stress and the pathogenesis of Alzheimer's disease. Oxid Med Cell Longev 2013;2013:316523.
- Radi E, Formichi P, Battisti C, Federico A. Apoptosis and oxidative stress in neurodegenerative diseases. J Alzheimers Dis 2014;42:S125-S152.
- Matsuoka Y, Picciano M, La Francois J, Duff K. Fibrillar beta-amyloid evokes oxidative damage in a transgenic mouse model of Alzheimer's disease. Neuroscience 2001;104:609-613.
- Murakami K, Murata N, Noda Y, Tahara S, Kaneko T, Kinoshita N, Hatsuta H, Murayama S, Barnham KJ, Irie K, Shirasawa T, Shimizu T. SOD1 (copper/zinc superoxide dismutase) deficiency drives amyloid beta protein oligomerization and memory loss in mouse model of Alzheimer disease. J Biol Chem 2011;286:44557-44568.
- Chen L, Na R, Gu M, Richardson A, Ran Q. Lipid peroxidation up-regulates BACE1 expression in vivo: a possible early event of amyloidogenesis in Alzheimer's disease. J Neurochem 2008;107:197-207.
- Stamer K, Vogel R, Thies E, Mandelkow E, Mandelkow EM. Tau blocks traffic of organelles, neurofilaments, and APP vesicles in neurons and enhances oxidative stress. J Cell Biol 2002;156:1051-1063.
- Yoshiyama Y, Higuchi M, Zhang B, Huang SM, Iwata N, Saido TC, Maeda J, Suhara T, Trojanowski JQ, Lee VM. Synapse loss and microglial activation precede tangles in a P301S tauopathy mouse model. Neuron 2007;53:337-351.
- David DC, Hauptmann S, Scherping I, Schuessel K, Keil U, Rizzu P, Ravid R, Dröse S, Brandt U, Muller WE, Eckert A, Götz J. Proteomic and functional analyses reveal a mitochondrial dysfunction in P301L tau transgenic mice. J Biol Chem 2005;280:23802-23814.
- Halverson RA, Lewis J, Frausto S, Hutton M, Muma NA. Tau protein is cross-linked by transglutaminase in P301L tau transgenic mice. J Neurosci 2005;25:1226-1233.
- Picone P, Nuzzo D, Caruana L, Scafidi V, Di Carlo M. Mitochondrial dysfunction: different routes to Alzheimer's disease therapy. Oxid Med Cell Longev 2014;2014:780179.
- Castellani R, Hirai K, Aliev G, Drew KL, Nunomura A, Takeda A, Cash AD, Obrenovich ME, Perry G, Smith MA. Role of mitochondrial dysfunction in Alzheimer's disease. J Neurosci Res 2002;70:357-360.
- Bubber P, Haroutunian V, Fisch G, Blass JP, Gibson GE. Mitochondrial abnormalities in Alzheimer brain: mechanistic implications. Ann Neurol 2005;57:695-703.
- Ito E, Oka K, Etcheberrigaray R, Nelson TJ, McPhie DL, Tofel-Grehl B, Gibson GE, Alkon DL. Internal Ca2+ mobilization is altered in fibroblasts from patients with Alzheimer disease. Proc Natl Acad Sci U S A 1994;91:534-538.
- Moon HE, Paek SH. Mitochondrial Dysfunction in Parkinson's Disease. Exp Neurobiol 2015;24:103-116.
- Blesa J, Trigo-Damas I, Quiroga-Varela A, Jackson-Lewis VR. Oxidative stress and Parkinson's disease. Front Neuroanat 2015;9:91.
- Schapira AH. Mitochondria in the aetiology and pathogenesis of Parkinson's disease. Lancet Neurol 2008;7:97-109.
- Franco-Iborra S, Vila M, Perier C. The Parkinson disease mitochondrial hypothesis: where are we at?. Neuroscientist
- Hauser DN, Hastings TG. Mitochondrial dysfunction and oxidative stress in Parkinson's disease and monogenic parkinsonism. Neurobiol Dis 2013;51:35-42.
- Swerdlow RH, Parks JK, Miller SW, Tuttle JB, Trimmer PA, Sheehan JP, Bennett JP, Davis RE, Parker WD. Origin and functional consequences of the complex I defect in Parkinson's disease. Ann Neurol 1996;40:663-671.
- Bandmann O, Sweeney MG, Daniel SE, Marsden CD, Wood NW. Mitochondrial DNA polymorphisms in pathologically proven Parkinson's disease. J Neurol 1997;244:262-265.
- Sian J, Dexter DT, Lees AJ, Daniel S, Agid Y, Javoy-Agid F, Jenner P, Marsden CD. Alterations in glutathione levels in Parkinson's disease and other neurodegenerative disorders affecting basal ganglia. Ann Neurol 1994;36:348-355.
- Dexter DT, Wells FR, Lees AJ, Agid F, Agid Y, Jenner P, Marsden CD. Increased nigral iron content and alterations in other metal ions occurring in brain in Parkinson's disease. J Neurochem 1989;52:1830-1836.
- Oakley AE, Collingwood JF, Dobson J, Love G, Perrott HR, Edwardson JA, Elstner M, Morris CM. Individual dopaminergic neurons show raised iron levels in Parkinson disease. Neurology 2007;68:1820-1825.
- Lovell MA, Ehmann WD, Butler SM, Markesbery WR. Elevated thiobarbituric acid-reactive substances and antioxidant enzyme activity in the brain in Alzheimer's disease. Neurology 1995;45:1594-1601.
- Lovell MA, Xie C, Markesbery WR. Decreased glutathione transferase activity in brain and ventricular fluid in Alzheimer's disease. Neurology 1998;51:1562-1566.
- Rinaldi P, Polidori MC, Metastasio A, Mariani E, Mattioli P, Cherubini A, Catani M, Cecchetti R, Senin U, Mecocci P. Plasma antioxidants are similarly depleted in mild cognitive impairment and in Alzheimer's disease. Neurobiol Aging 2003;24:915-919.
- Sinclair AJ, Bayer AJ, Johnston J, Warner C, Maxwell SR. Altered plasma antioxidant status in subjects with Alzheimer's disease and vascular dementia. Int J Geriatr Psychiatry 1998;13:840-845.
- McCaddon A, Hudson P, Hill D, Barber J, Lloyd A, Davies G, Regland B. Alzheimer's disease and total plasma aminothiols. Biol Psychiatry 2003;53:254-260.
- Kalra J, Rajput AH, Mantha SV, Prasad K. Serum antioxidant enzyme activity in Parkinson's disease. Mol Cell Biochem 1992;110:165-168.
- Ilic TV, Jovanovic M, Jovicic A, Tomovic M. Oxidative stress indicators are elevated in de novo Parkinson's disease patients. Funct Neurol 1999;14:141-147.
- Kocatürk PA, Akbostanci MC, Tan F, Kavas GO. Superoxide dismutase activity and zinc and copper concentrations in Parkinson's disease. Pathophysiology 2000;7:63-67.
- Serra JA, Dominguez RO, de Lustig ES, Guareschi EM, Famulari AL, Bartolome EL, Marschoff ER. Parkinson's disease is associated with oxidative stress: comparison of peripheral antioxidant profiles in living Parkinson's, Alzheimer's and vascular dementia patients. J Neural Transm 2001;108:1135-1148.
- Younes-Mhenni S, Frih-Ayed M, Kerkeni A, Bost M, Chazot G. Peripheral blood markers of oxidative stress in Parkinson's disease. Eur Neurol 2007;58:78-83.
- Matsuzawa D, Hashimoto K. Magnetic resonance spectroscopy study of the antioxidant defense system in schizophrenia. Antioxid Redox Signal 2011;15:2057-2065.
- Govindaraju V, Young K, Maudsley AA. Proton NMR chemical shifts and coupling constants for brain metabolites. NMR Biomed 2000;13:129-153.
- Terpstra M, Henry PG, Gruetter R. Measurement of reduced glutathione (GSH) in human brain using LCModel analysis of difference-edited spectra. Magn Reson Med 2003;50:19-23.
- Mandal PK, Tripathi M, Sugunan S. Brain oxidative stress: detection and mapping of anti-oxidant marker 'Glutathione' in different brain regions of healthy male/female, MCI and Alzheimer patients using non-invasive magnetic resonance spectroscopy. Biochem Biophys Res Commun 2012;417:43-48.
- Terpstra M, Gruetter R. 1H NMR detection of vitamin C in human brain in vivo. Magn Reson Med 2004;51:225-229.
- Mandal PK, Saharan S, Tripathi M, Murari G. Brain glutathione levels - a novel biomarker for mild cognitive impairment and Alzheimer's Disease. Biol Psychiatry
- Shih YY, Büchert M, Chung HW, Hennig J, von Elverfeldt D. Vitamin C estimation with standard (1)H spectroscopy using a clinical 3T MR system: detectability and reliability within the human brain. J Magn Reson Imaging 2008;28:351-358.
- Ikawa M, Okazawa H, Tsujikawa T, Matsunaga A, Yamamura O, Mori T, Hamano T, Kiyono Y, Nakamoto Y, Yoneda M. Increased oxidative stress is related to disease severity in the ALS motor cortex: a PET study. Neurology 2015;84:2033-2039.
- Ikawa M, Okazawa H, Kudo T, Kuriyama M, Fujibayashi Y, Yoneda M. Evaluation of striatal oxidative stress in patients with Parkinson's disease using [62Cu]ATSM PET. Nucl Med Biol 2011;38:945-951.
- Fujibayashi Y, Taniuchi H, Yonekura Y, Ohtani H, Konishi J, Yokoyama A. Copper-62-ATSM: a new hypoxia imaging agent with high membrane permeability and low redox potential. J Nucl Med 1997;38:1155-1160.
- Obata A, Yoshimi E, Waki A, Lewis JS, Oyama N, Welch MJ, Saji H, Yonekura Y, Fujibayashi Y. Retention mechanism of hypoxia selective nuclear imaging/radiotherapeutic agent cu-diacetyl-bis(N4-methylthiosemicarbazone) (Cu-ATSM) in tumor cells. Ann Nucl Med 2001;15:499-504.
- Hawkins CL, Davies MJ. Detection and characterisation of radicals in biological materials using EPR methodology. Biochim Biophys Acta 2014;1840:708-721.
- Lee MC. Assessment of oxidative stress and antioxidant property using electron spin resonance (ESR) spectroscopy. J Clin Biochem Nutr 2013;52:1-8.
- Mrakic-Sposta S, Gussoni M, Montorsi M, Porcelli S, Vezzoli A. Assessment of a standardized ROS production profile in humans by electron paramagnetic resonance. Oxid Med Cell Longev 2012;2012:973927.
- Ienco EC, LoGerfo A, Carlesi C, Orsucci D, Ricci G, Mancuso M, Siciliano G. Oxidative stress treatment for clinical trials in neurodegenerative diseases. J Alzheimers Dis 2011;24:111-126.
- Saso L, Firuzi O. Pharmacological applications of antioxidants: lights and shadows. Curr Drug Targets 2014;15:1177-1199.
- Morris MC, Beckett LA, Scherr PA, Hebert LE, Bennett DA, Field TS, Evans DA. Vitamin E and vitamin C supplement use and risk of incident Alzheimer disease. Alzheimer Dis Assoc Disord 1998;12:121-126.
- Morris MC, Evans DA, Bienias JL, Tangney CC, Bennett DA, Aggarwal N, Wilson RS, Scherr PA. Dietary intake of antioxidant nutrients and the risk of incident Alzheimer disease in a biracial community study. JAMA 2002;287:3230-3237.
- Zandi PP, Anthony JC, Khachaturian AS, Stone SV, Gustafson D, Tschanz JT, Norton MC, Welsh-Bohmer KA, Breitner JC, Cache County Study Group. Reduced risk of Alzheimer disease in users of antioxidant vitamin supplements: the Cache County Study. Arch Neurol 2004;61:82-88.
- Sano M, Ernesto C, Thomas RG, Klauber MR, Schafer K, Grundman M, Woodbury P, Growdon J, Cotman CW, Pfeiffer E, Schneider LS, Thal LJ, The Alzheimer's Disease Cooperative Study. A controlled trial of selegiline, alpha-tocopherol, or both as treatment for Alzheimer's disease. N Engl J Med 1997;336:1216-1222.
- Petersen RC, Thomas RG, Grundman M, Bennett D, Doody R, Ferris S, Galasko D, Jin S, Kaye J, Levey A, Pfeiffer E, Sano M, van Dyck CH, Thal LJ, Alzheimer's Disease Cooperative Study Group. Vitamin E and donepezil for the treatment of mild cognitive impairment. N Engl J Med 2005;352:2379-2388.
- Dysken MW, Sano M, Asthana S, Vertrees JE, Pallaki M, Llorente M, Love S, Schellenberg GD, McCarten JR, Malphurs J, Prieto S, Chen P, Loreck DJ, Trapp G, Bakshi RS, Mintzer JE, Heidebrink JL, Vidal-Cardona A, Arroyo LM, Cruz AR, Zachariah S, Kowall NW, Chopra MP, Craft S, Thielke S, Turvey CL, Woodman C, Monnell KA, Gordon K, Tomaska J, Segal Y, Peduzzi PN, Guarino PD. Effect of vitamin E and memantine on functional decline in Alzheimer disease: the TEAM-AD VA cooperative randomized trial. JAMA 2014;311:33-44.
- Galasko DR, Peskind E, Clark CM, Quinn JF, Ringman JM, Jicha GA, Cotman C, Cottrell B, Montine TJ, Thomas RG, Aisen P, Alzheimer's Disease Cooperative Study. Antioxidants for Alzheimer disease: a randomized clinical trial with cerebrospinal fluid biomarker measures. Arch Neurol 2012;69:836-841.
- Adair JC, Knoefel JE, Morgan N. Controlled trial of N-acetylcysteine for patients with probable Alzheimer's disease. Neurology 2001;57:1515-1517.
- Bergamasco B, Scarzella L, La Commare P. Idebenone, a new drug in the treatment of cognitive impairment in patients with dementia of the Alzheimer type. Funct Neurol 1994;9:161-168.
- Weyer G, Babej-Dölle RM, Hadler D, Hofmann S, Herrmann WM. A controlled study of 2 doses of idebenone in the treatment of Alzheimer's disease. Neuropsychobiology 1997;36:73-82.
- Gutzmann H, Hadler D. Sustained efficacy and safety of idebenone in the treatment of Alzheimer's disease: update on a 2-year double-blind multicentre study. J Neural Transm Suppl 1998;54:301-310.
- Thal LJ, Grundman M, Berg J, Ernstrom K, Margolin R, Pfeiffer E, Weiner MF, Zamrini E, Thomas RG. Idebenone treatment fails to slow cognitive decline in Alzheimer's disease. Neurology 2003;61:1498-1502.
- Zhang SM, Hernan MA, Chen H, Spiegelman D, Willett WC, Ascherio A. Intakes of vitamins E and C, carotenoids, vitamin supplements, and PD risk. Neurology 2002;59:1161-1169.
- Parkinson Study Group. Effects of tocopherol and deprenyl on the progression of disability in early Parkinson's disease. N Engl J Med 1993;328:176-183.
- Shoulson I, Parkinson Study Group. Deprenyl and tocopherol antioxidative therapy of parkinsonism (DATATOP). Acta Neurol Scand Suppl 1989;126:171-175.
- Shults CW, Oakes D, Kieburtz K, Beal MF, Haas R, Plumb S, Juncos JL, Nutt J, Shoulson I, Carter J, Kompoliti K, Perlmutter JS, Reich S, Stern M, Watts RL, Kurlan R, Molho E, Harrison M, Lew M, Parkinson Study Group. Effects of coenzyme Q10 in early Parkinson disease: evidence of slowing of the functional decline. Arch Neurol 2002;59:1541-1550.
- Beal MF, Oakes D, Shoulson I, Henchcliffe C, Galpern WR, Haas R, Juncos JL, Nutt JG, Voss TS, Ravina B, Shults CM, Helles K, Snively V, Lew MF, Griebner B, Watts A, Gao S, Pourcher E, Bond L, Kompoliti K, Agarwal P, Sia C, Jog M, Cole L, Sultana M, Kurlan R, Richard I, Deeley C, Waters CH, Figueroa A, Arkun A, Brodsky M, Ondo WG, Hunter CB, Jimenez-Shahed J, Palao A, Miyasaki JM, So J, Tetrud J, Reys L, Smith K, Singer C, Blenke A, Russell DS, Cotto C, Friedman JH, Lannon M, Zhang L, Drasby E, Kumar R, Subramanian T, Ford DS, Grimes DA, Cote D, Conway J, Siderowf AD, Evatt ML, Sommerfeld B, Lieberman AN, Okun MS, Rodriguez RL, Merritt S, Swartz CL, Martin WR, King P, Stover N, Guthrie S, Watts RL, Ahmed A, Fernandez HH, Winters A, Mari Z, Dawson TM, Dunlop B, Feigin AS, Shannon B, Nirenberg MJ, Ogg M, Ellias SA, Thomas CA, Frei K, Bodis-Wollner I, Glazman S, Mayer T, Hauser RA, Pahwa R, Langhammer A, Ranawaya R, Derwent L, Sethi KD, Farrow B, Prakash R, Litvan I, Robinson A, Sahay A, Gartner M, Hinson VK, Markind S, Pelikan M, Perlmutter JS, Hartlein J, Molho E, Evans S, Adler CH, Duffy A, Lind M, Elmer L, Davis K, Spears J, Wilson S, Leehey MA, Hermanowicz N, Niswonger S, Shill HA, Obradov S, Rajput A, Cowper M, Lessig S, Song D, Fontaine D, Zadikoff C, Williams K, Blindauer KA, Bergholte J, Propsom CS, Stacy MA, Field J, Mihaila D, Chilton M, Uc EY, Sieren J, Simon DK, Kraics L, Silver A, Boyd JT, Hamill RW, Ingvoldstad C, Young J, Thomas K, Kostyk SK, Wojcieszek J, Pfeiffer RF, Panisset M, Beland M, Reich SG, Cines M, Zappala N, Rivest J, Zweig R, Lumina LP, Hilliard CL, Grill S, Kellermann M, Tuite P, Rolandelli S, Kang UJ, Young J, Rao J, Cook MM, Severt L, Boyar K, Parkinson Study Group QE3 Investigators. A randomized clinical trial of high-dosage coenzyme Q10 in early Parkinson disease: no evidence of benefit. JAMA Neurol 2014;71:543-552.
- Murphy MP. Antioxidants as therapies: can we improve on nature?. Free Radic Biol Med 2014;66:20-23.
- Firuzi O, Miri R, Tavakkoli M, Saso L. Antioxidant therapy: current status and future prospects. Curr Med Chem 2011;18:3871-3888.
- Praticò D. Evidence of oxidative stress in Alzheimer's disease brain and antioxidant therapy: lights and shadows. Ann N Y Acad Sci 2008;1147:70-78.