Articles

Article Tools
 View Full Text View Full Text |
 Abstract Abstract |
 Article as PDF Article as PDF |
 Print this Article Print this Article |
 Pubmed Pubmed |
 PMC PMC |
 PubReader PubReader |
 Export to Citation Export to Citation |
 Email Alerts Email Alerts |
 Open Access Open Access |
Share this article on :



Stats or Metrics
Article
Original Article
Exp Neurobiol 2016; 25(3): 113-119
Published online June 30, 2016
https://doi.org/10.5607/en.2016.25.3.113
© The Korean Society for Brain and Neural Sciences
Non-cell-autonomous Neurotoxicity of α-synuclein Through Microglial Toll-like Receptor 2
Changyoun Kim1,2, He-Jin Lee3, Eliezer Masliah2 and Seung-Jae Lee1*
1Department of Biomedical Sciences, Neuroscience Research Institute, Seoul National University College of Medicine, Seoul 03080, Korea, 2Departments of Neurosciences and Pathology, School of Medicine, University of California, San Diego, La Jolla, CA 92093, USA, 3Department of Anatomy, School of Medicine, Konkuk University, Seoul 05030, Korea
Correspondence to: *To whom correspondence should be addressed.
TEL: 82-2-3668-7037, FAX: 82-2-447-5683
e-mail: sjlee66@snu.ac.kr
Synucleinopathies are a collection of neurological diseases that are characterized by deposition of α-synuclein aggregates in neurons and glia. These diseases include Parkinson's disease (PD), dementia with Lewy bodies, and multiple system atrophy. Although it has been increasingly clear that α-synuclein is implicated in the pathogenesis of PD and other synucleinopathies, the precise mechanism underlying the disease process remains to be unraveled. The past studies on how α-synuclein exerts pathogenic actions have focused on its direct, cell-autonomous neurotoxic effects. However, recent findings suggested that there might be indirect, non-cell-autonomous pathways, perhaps through the changes in glial cells, for the pathogenic actions of this protein. Here, we present evidence that α-synuclein can cause neurodegeneration through a non-cell-autonomous manner. We show that α-synuclein can be secreted from neurons and induces inflammatory responses in microglia, which in turn secreted neurotoxic agents into the media causing neurodegeneration. The neurotoxic response of microglia was mediated by activation of toll-like receptor 2 (TLR2), a receptor for neuron-derived α-synuclein. This work suggests that TLR2 is the key molecule that mediates non-cell-autonomous neurotoxic effects of α-synuclein, hence a candidate for the therapeutic target.
Keywords: Parkinson’s disease, α-synuclein, Toll-like receptor 2, Microglia, Neurodegeneration
INTRODUCTION
Abnormal deposition of α-synuclein is a key pathological feature of α-synucleinopathies, such as Parkinson's disease (PD) [1,2]. Past studies have established strong relationship between α-synuclein aggregation and disease onset and progression of PD [3,4]. Several mechanisms have been suggested as to how accumulation of α-synuclein induced neurodegeneration. These include inhibition of vesicle recycling, blocking of endoplasmic reticulum transport, impairment of mitochondrial energy production, and disruption of protein degradation process [3]. These mechanisms assume cell-autonomous pathogenic actions of α-synuclein in neurons. On the other hand, non-cell-autonomous actions of α-synuclein through modulation of glial cells has only begun to attract interests.
α-synuclein is a typical neuronal cytosolic protein; however, recent studies revealed the presence of some α-synuclein in extracellular space, such as cerebrospinal fluid, serum, and brain interstitial fluid [5,6,7]. Various forms of unconventional exocytosis, such as exosome-associated exocytosis and exophagy, have been suggested as mechanisms of neuronal α-synuclein secretion [8]. When secreted, extracellular α-synuclein can be transferred to neighboring neurons and glia, inducing cytotoxicity and pathological propagation in neurons [9,10] and causing neuroinflammation in glial cells [11,12].
Neuroinflammation is one of the key pathological features of many neurodegenerative diseases including PD [13]. Microglia, a brain resident immune cell, plays central role in the process of neuroinflammation [13,14,15]. Microglia can be activated by various types of stimuli, including brain injury, ischemia, and inflammatory stimuli [16]. When activated, microglia produces proinflammatory cytokines, chemokines, intracellular reactive oxygen species (iROS), and nitric oxide (NO), and therefore, chronic activation of miroglia could create a microenvironment where neurodegeneration is favored [17,18].
Although awareness of importance of glial changes is increasing, the mechanism of glial activation in the pathogenic processes remains largely unknown. Our previous study showed that neuron-released α-synuclein triggered pro-inflammatory activation of microglia through the interaction with TLR2 [12]. Herein, we investigated the effects of α-synuclein-induced microglial activation on neurodegeneration and the role of TLR2 in the microglia-mediated neurotoxicity.
MATERIALS AND METHODS
Sprague-Dawley rats and C57BL/6 mice were obtained from Samtako (Osan, Korea). TLR2-deficient mice were purchased from Oriental Bioservice (Kyoto, Japan) [19]. The animal use protocol was approved by Konkuk University's Animal Care and Use Committee. Genotypes of mice were analyzed by PCR using the following primers; for wild type TLR2: a + b and for TLR2-deficiend (Neo): b + c. a: 5'-GTT TAG TGC CTG TAT CCA GTC AGT GCG-3'. b: 5'-TTG GAT AAG TCT GAT AGC CTT GCC TCC-3'. c: 5'-ATC GCC TTC TAT CGC CTT CTT GAC GAG-3'.
All-trans retinoic acid and Lipopolysaccharides (LPS) were purchased from Sigma Aldrich (St. Louis, MO). Maintenance and differentiation of SH-SY5Y human neuroblastoma has been previously described [20]. Rat and mouse primary neuron and microglia were cultured as described previously [12].
Preparation of neuronal α-synuclein conditioned media (αSCM) and β-galactosidase conditioned media (LZCM) have been previously described [12]. As previously reported, αSCM contains l.06 ± 0.371 µg/ml of α-synuclein [12]. In this study, primary neurons and microglia were treated with approximate 5.3 µg/ml of α-synuclein (Fig. 1A).
To generate microglial conditioned medium (MgCM), rat and mouse primary microglia were treated with DMEM, LZCM, αSCM, or LPS (1 µg/ml) for 1 hour, then washed for 4 times with phosphate buffered saline. After a 6-hour post-incubation with fresh neurobasal medium, the culture medium were collected and centrifuged to remove cell debris at 10,000 × g for 10 minutes.
Three different assays have been used to determine neurotoxicity in this study; axonal bleb analysis, neuronal cell body counting, and cell viability assay. Briefly, rat cortical primary neurons were treated with DMEM, LZCM, αSCM, LPS (1 µg/ml), or various types of MgCM for 24 hours. The numbers of axonal blebs were counted in 50 randomly chosen neurons for each experiment. Total numbers of neuronal cell body were analyzed in 10 randomly chosen areas (1 mm2) for each experiment. Microscopic analyses of neurons were performed using FV10-ASE 1.7 software (Olympus, Tokyo, Japan). Cell viability was determined by CyQUANT cell proliferation assay kit (Invitrogen, Carlsbad, CA) according to the manufacturer's instruction.
InStat (GraphPad) was used for all statistical analyses. Statistical significances of data were determined using one-way ANOVA. All data are presented as means ± s.e.m.
To investigate the non-cell-autonomous neurotoxic effects of α-synuclein, we generated conditioned media from resting and activated primary microglia after pre-treating these cells with neuronal conditioned media. Rat primary microglia were treated with DMEM, conditioned medium from LacZ-expressing differentiated SH-SY5Y cells (LZCM), conditioned medium from α-synuclein-expressing differentiated SH-SY5Y cells (αSCM), or LPS (1 µg/ml). Conditioned media from the pre-treated microglia were named DMEM-MgCM, LZCM-MgCM, αSCM-MgCM, and LPS-MgCM, respectively. These MgCMs were treated to rat primary neurons for 24 hours (MgCM+). As controls, neurons were directly treated with DMEM, LZCM, αSCM, or LPS (MgCM-) (Fig. 1A).
Neurodegeneration was determined by the formation of axonal bleb and the loss of neuronal cell bodies. Direct treatment of neuron-released α-synuclein slightly increased the numbers of axonal bleb (Fig. 1B, C) and the loss of cell bodies (Fig. 1B, D), while treatments of DMEM and LZCM did not induce cytotoxicity (Fig. 1B~D). On the other hand, treatment of MgCM from αSCM-exposed microglia strikingly increased the numbers of axonal bleb formation (Fig. 1B, C) and the loss of neuronal cell bodies (Fig. 1B, D). Treatment of LPS-MgCM also clearly increased the numbers of axonal bleb and the cell body loss. These results suggest that microglia produces and secrets neurotoxic agents in response to extracellular α-synuclein exposure and LPS.
Our previous study demonstrated that oligomeric forms of neuron-released α-synuclein can be recognized by TLR2 on the surface of microglia, thereby inducing microglia activation though the TLR2 signaling cascade [12]. To validate the role of TLR2 in αSCM-induced microglial neurotoxicity, we generated MgCMs using wild-type (WT-mMgCM) and Tlr2-deficient (Tlr2-/--mMgCM) mice microglia. Primary mouse microglia were treated with LZCM, αSCM, or LPS (LZCM-mMgCM, αSCM-mMgCM, or LPS-mMgCM), then the medium were treated to the rat primary neurons for 24 hours. As controls, LZCM, αSCM, and LPS were directly treated to the rat primary neurons. Similar to the results obtained from rat MgCMs, mMgCMs acquired from wild-type mice microglia significantly increased the formations of axonal bleb and cytotoxicity after exposed to αSCM and LPS (Fig. 2B-D). In contrast, LZCM-mMgCM from both wild-type and Tlr2-/- mice did not show neurotoxic effect (Fig. 2B-D). Interestingly, the microglial neurotoxicity induced by αSCM exposure was completely abrogated by Tlr2 gene depletion, while LPS-induced microglial neurotoxicity was not affected (Fig. 2B-D). These results suggest that neuron-released α-synuclein can exert its neurotoxic effects in a non-cell-autonomous manner through the activation of microglial TLR2.
Most studies have focused on the direct, cell-autonomous neurotoxic effects of neuronal α-synuclein. Recently, however, discovery of extracellular α-synuclein and increased interests in glial contribution to PD pathogenesis prompted us to investigate the role of microglia in non-cell-autonomous neurotoxic action of α-synuclein and the mechanism of the action [21]. In the present study, we show indirect mechanism of neurotoxicity of neuronal α-synuclein, in which neuron-secreted α-synuclein activates microglia leading to the secretion of neurotoxic agents from microglia in a TLR2-dependent manner. Neuron-released α-synuclein indeed showed a slight neurotoxicity on its own. However, the indirect toxic pathway through microglial activation produced much bigger neurotoxic effect than the direct toxic pathway. Therefore, these results suggest that microglia may amplify the neurotoxicity of extracellular α-synuclein through triggering neuroinflammation processes.
It remains to be determined which secreted microglial agents mediate neurotoxic effects of activated microglia. Our previous study demonstrated that microglia exposed to neuron-released extracellular α-synuclein produced iROS, NO, and various types of cytokines, such as TNFα, IL-1β, and IL-6 [12]. Considering the fact that the microglial neurotoxicity can be transferable via conditioned medium, it is unlikely that such labile molecules as ROS are responsible for the toxicity. It would be more reasonable to assume that the secreted cytokines or other macromolecules mediate the toxic effects of microglia. Furthermore, studies have shown that microglia-derived inflammatory molecules, such as TNFα, IL-1β, IL-6, and NO contributed to the acceleration of dopamine neuron degeneration [22,23,24]. Determination of the microglial toxic agents that are secreted upon α-synuclein treatment would be an important next step towards identification of the therapeutic target.
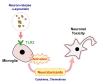
TLR2 is a key innate immune receptor belong to pattern recognition receptors [25]. Recent studies have revealed the relation between TLR2 and PD [12,26]. Studies with post-mortem brains have shown an elevation of microglial TLR2 expression in striatum, hippocampus, and substantia nigra of PD patients [27,28]. In addition to PD patients, the expression of microglial TLR2 was increased in an animal model of PD overexpressing human α-synuclein [28,29]. Our previous study suggested that TLR2 was the receptor for neuron-released oligomeric α-synuclein and mediated microglial inflammatory responses [12]. As a result of the activation, microglia produced neurotoxic byproducts, such as proinflammatory cytokines, NO, and iROS [12]. These effects were eliminated when α-synuclein was removed from the neuronal conditioned medium, indicating that α-synuclein was the principal substance eliciting the inflammatory responses through TLR2. In the current study, we also experimentally validated non-cell-autonomous neurotoxicity of neuron-released α-synuclein through TLR2-dependent microglial activation. Previous studies had also suggested TLR-mediated microglial activation [30,31]. However, our current study is different from the previous ones in two important aspects. First, the previous studies investigated the roles of TLRs in proinflammatory responses of microglia, whereas our current study went on to the next step and showed the neurotoxic consequence of TLR-mediated microglial activation. The aforementioned studies did not examined neurotoxicity of activated microglia. Second, the previous studies used recombinant α-synuclein proteins, while our study used natural α-synuclein proteins secreted from neuronal cells. The latter point is important because we had shown that neuron-secreted α-synuclein interacts only with TLR2 but not with TLR3 or TLR4, and that recombinant α-synuclein fibrils are not capable of activating TLR2 [12], suggesting that the recombinant protein acts differently from the natural neuron-derived α-synuclein. Therefore, our current study does not overlap with the previous two studies. Furthermore, recent study suggested that neuronal TLR2 regulates autophagy in neurons in such a way that TLR2 activation inhibits autophagy leading to accumulation of α-synuclein aggregates [26]. Therefore, blocking TLR2 function might be a potential approach towards developing therapy for PD (Fig. 3). In conclusion, we demonstrated that neuronal α-synuclein can exert its neurotoxic effects through the indirect, non-cell-autonomous manner, which involves activation of microglia via TLR2 activation.

{kind=link}

{kind=link}
{kind=link}
- McCann H, Stevens CH, Cartwright H, Halliday GM. Alpha-synucleinopathy phenotypes. Parkinsonism Relat Disord 2014;20:S62-S67.

- Lashuel HA, Overk CR, Oueslati A, Masliah E. The many faces of alpha-synuclein: from structure and toxicity to therapeutic target. Nat Rev Neurosci 2013;14:38-48.
- Cookson MR. Alpha-synuclein and neuronal cell death. Mol Neurodegener 2009;4:9.
- Abstracts of the 25th Annual Meeting of the American Society for Bone and Mineral Research. Minneapolis, Minnesota, USA. September 19-23, 2003. J Bone Miner Res 2003;18:S2-S463.
- Mollenhauer B, El-Agnaf OM, Marcus K, Trenkwalder C, Schlossmacher MG. Quantification of alpha-synuclein in cerebrospinal fluid as a biomarker candidate: review of the literature and considerations for future studies. Biomarkers Med 2010;4:683-699.
- Emmanouilidou E, Elenis D, Papasilekas T, Stranjalis G, Gerozissis K, Ioannou PC, Vekrellis K. Assessment of alpha-synuclein secretion in mouse and human brain parenchyma. PLoS One 2011;6:e22225.
- Lee HJ, Patel S, Lee SJ. Intravesicular localization and exocytosis of alpha-synuclein and its aggregates. J Neurosci 2005;25:6016-6024.
- Jang A, Lee HJ, Suk JE, Jung JW, Kim KP, Lee SJ. Non-classical exocytosis of alpha-synuclein is sensitive to folding states and promoted under stress conditions. J Neurochem 2010;113:1263-1274.
- Desplats P, Lee HJ, Bae EJ, Patrick C, Rockenstein E, Crews L, Spencer B, Masliah E, Lee SJ. Inclusion formation and neuronal cell death through neuron-to-neuron transmission of alpha-synuclein. Proc Natl Acad Sci U S A 2009;106:13010-13015.
- Danzer KM, Ruf WP, Putcha P, Joyner D, Hashimoto T, Glabe C, Hyman BT, McLean PJ. Heat-shock protein 70 modulates toxic extracellular alpha-synuclein oligomers and rescues trans-synaptic toxicity. FASEB J 2011;25:326-336.
- Lee HJ, Suk JE, Patrick C, Bae EJ, Cho JH, Rho S, Hwang D, Masliah E, Lee SJ. Direct transfer of alpha-synuclein from neuron to astroglia causes inflammatory responses in synucleinopathies. J Biol Chem 2010;285:9262-9272.
- Kim C, Ho DH, Suk JE, You S, Michael S, Kang J, Joong Lee S, Masliah E, Hwang D, Lee HJ, Lee SJ. Neuron-released oligomeric alpha-synuclein is an endogenous agonist of TLR2 for paracrine activation of microglia. Nat Commun 2013;4:1562.
- Wake H, Moorhouse AJ, Miyamoto A, Nabekura J. Microglia: actively surveying and shaping neuronal circuit structure and function. Trends Neurosci 2013;36:209-217.
- Carson MJ. Microglia as liaisons between the immune and central nervous systems: functional implications for multiple sclerosis. Glia 2002;40:218-231.
- Jeong HK, Ji K, Min K, Joe EH. Brain inflammation and microglia: facts and misconceptions. Exp Neurobiol 2013;22:59-67.
- Hanisch UK, Kettenmann H. Microglia: active sensor and versatile effector cells in the normal and pathologic brain. Nat Neurosci 2007;10:1387-1394.
- Long-Smith CM, Sullivan AM, Nolan YM. The influence of microglia on the pathogenesis of Parkinson's disease. Prog Neurobiol 2009;89:277-287.
- Woo JH, Lee JH, Kim H, Park SJ, Joe EH, Jou I. Control of inflammatory responses: a new paradigm for the treatment of chronic neuronal diseases. Exp Neurobiol 2015;24:95-102.
- Takeuchi O, Hoshino K, Kawai T, Sanjo H, Takada H, Ogawa T, Takeda K, Akira S. Differential roles of TLR2 and TLR4 in recognition of gram-negative and gram-positive bacterial cell wall components. Immunity 1999;11:443-451.
- Bae EJ, Lee C, Lee HJ, Kim S, Lee SJ. ATP13A2/PARK9 deficiency neither cause lysosomal impairment nor alter alpha-synuclein metabolism in SH-SY5Y cells. Exp Neurobiol 2014;23:365-371.
- Block ML, Hong JS. Microglia and inflammation-mediated neurodegeneration: multiple triggers with a common mechanism. Prog Neurobiol 2005;76:77-98.
- Ferrari CC, Pott Godoy MC, Tarelli R, Chertoff M, Depino AM, Pitossi FJ. Progressive neurodegeneration and motor disabilities induced by chronic expression of IL-1beta in the substantia nigra. Neurobiol Dis 2006;24:183-193.
- McCoy MK, Martinez TN, Ruhn KA, Szymkowski DE, Smith CG, Botterman BR, Tansey KE, Tansey MG. Blocking soluble tumor necrosis factor signaling with dominant-negative tumor necrosis factor inhibitor attenuates loss of dopaminergic neurons in models of Parkinson’s disease. J Neurosci 2006;26:9365-9375.
- Davalos D, Grutzendler J, Yang G, Kim JV, Zuo Y, Jung S, Littman DR, Dustin ML, Gan WB. ATP mediates rapid microglial response to local brain injury in vivo. Nat Neurosci 2005;8:752-758.
- Okun E, Griffioen KJ, Mattson MP. Toll-like receptor signaling in neural plasticity and disease. Trends Neurosci 2011;34:269-281.
- Kim C, Rockenstein E, Spencer B, Kim HK, Adame A, Trejo M, Stafa K, Lee HJ, Lee SJ, Masliah E. Antagonizing neuronal toll-like receptor 2 prevents synucleinopathy by activating autophagy. Cell Reports 2015;13:771-782.
- Doorn KJ, Moors T, Drukarch B, van de Berg WD, Lucassen PJ, van Dam AM. Microglial phenotypes and toll-like receptor 2 in the substantia nigra and hippocampus of incidental Lewy body disease cases and Parkinson's disease patients. Acta Neuropathol Commun 2014;2:90.
- Drouin-Ouellet J, St-Amour I, Saint-Pierre M, Lamontagne-Proulx J, Kriz J, Barker RA, Cicchetti F. Toll-like receptor expression in the blood and brain of patients and a mouse model of Parkinson's disease. Int J Neuropsychopharmacol 2014;18:pyu103.
- Letiembre M, Liu Y, Walter S, Hao W, Pfander T, Wrede A, Schulz-Schaeffer W, Fassbender K. Screening of innate immune receptors in neurodegenerative diseases: a similar pattern. Neurobiol Aging 2009;30:759-768.
- Béraud D, Maguire-Zeiss KA. Misfolded alphasynuclein and toll-like receptors: therapeutic targets for Parkinsons disease. Parkinsonism Relat Disord 2012;18:S17-S20.
- Roodveldt C, Labrador-Garrido A, Gonzalez-Rey E, Lachaud CC, Guilliams T, Fernandez-Montesinos R, Benitez-Rondan A, Robledo G, Hmadcha A, Delgado M, Dobson CM, Pozo D. Preconditioning of microglia by alpha-synuclein strongly affects the response induced by toll-like receptor (TLR) stimulation. PLoS One 2013;8:e79160.