Articles

Article Tools
 View Full Text View Full Text |
 Abstract Abstract |
 Article as PDF Article as PDF |
 Print this Article Print this Article |
 Pubmed Pubmed |
 PMC PMC |
 PubReader PubReader |
 Export to Citation Export to Citation |
 Email Alerts Email Alerts |
 Open Access Open Access |
Share this article on :



Stats or Metrics
Article
Review Article
Exp Neurobiol 2019; 28(5): 547-553
Published online October 31, 2019
https://doi.org/10.5607/en.2019.28.5.547
© The Korean Society for Brain and Neural Sciences
Targeting Microglial and Neuronal Toll-like Receptor 2 in Synucleinopathies
Somin Kwon, Michiyo Iba, Eliezer Masliah and Changyoun Kim*
Molecular Neuropathology Section, Laboratory of Neurogenetics, National Institute on Aging, National Institutes of Health, Bethesda, MD 20892, USA
Correspondence to: *To whom correspondence should be addressed.
TEL: 1-301-451-2120, FAX: 1-301-451-7295
e-mail: changyoun.kim@nih.gov
This is an Open Access article distributed under the terms of the Creative Commons Attribution Non-Commercial License(http://creativecommons.org/licenses/by-nc/4.0) which permits unrestricted non-commercial use, distribution, andreproduction in any medium, provided the original work is properly cited.
Abstract
- Go to
- Abstract
- Graphical Abstract
- INTRODUCTION
- PATHOLOGICAL INTERACTION OF MICROGLIAL TLR2 AND EXTRACELLULAR α-SYNUCLEIN
- PATHOLOGICAL INTERACTION OF NEURONAL TLR2 AND EXTRACELLULAR α-SYNUCLEIN
- TLR2 IN CELL-TO-CELL TRANSMISSION
- TLR2 IMMUNOTHERAPY IN SYNUCLEINOPATHY MOUSE MODEL
- CONCLUSIONS
- ACKNOWLEDGEMENTS
- Figure
- Reference
Synucleinopathies are neurodegenerative disorders characterized by the progressive accumulation of α-synuclein (α-syn) in neurons and glia and include Parkinson’s disease (PD) and dementia with Lewy bodies (DLB). In this review, we consolidate our key findings and recent studies concerning the role of Toll-like receptor 2 (TLR2), a pattern recognition innate immune receptor, in the pathogenesis of synucleinopathies. First, we address the pathological interaction of α-syn with microglial TLR2 and its neurotoxic inflammatory effects. Then, we show that neuronal TLR2 activation not only induces abnormal α-syn accumulation by impairing autophagy, but also modulates α-syn transmission. Finally, we demonstrate that administration of a TLR2 functional inhibitor improves the neuropathology and behavioral deficits of a synucleinopathy mouse model. Altogether, we present TLR2 modulation as a promising immunotherapy for synucleinopathies.
Graphical Abstract
- Go to
- Abstract
- Graphical Abstract
- INTRODUCTION
- PATHOLOGICAL INTERACTION OF MICROGLIAL TLR2 AND EXTRACELLULAR α-SYNUCLEIN
- PATHOLOGICAL INTERACTION OF NEURONAL TLR2 AND EXTRACELLULAR α-SYNUCLEIN
- TLR2 IN CELL-TO-CELL TRANSMISSION
- TLR2 IMMUNOTHERAPY IN SYNUCLEINOPATHY MOUSE MODEL
- CONCLUSIONS
- ACKNOWLEDGEMENTS
- Figure
- Reference
Keywords: Neuroinflammation, &alpha,-synuclein, Toll-like receptor 2, Immunotherapy, Synucleinopathy
INTRODUCTION
- Go to
- Abstract
- Graphical Abstract
- INTRODUCTION
- PATHOLOGICAL INTERACTION OF MICROGLIAL TLR2 AND EXTRACELLULAR α-SYNUCLEIN
- PATHOLOGICAL INTERACTION OF NEURONAL TLR2 AND EXTRACELLULAR α-SYNUCLEIN
- TLR2 IN CELL-TO-CELL TRANSMISSION
- TLR2 IMMUNOTHERAPY IN SYNUCLEINOPATHY MOUSE MODEL
- CONCLUSIONS
- ACKNOWLEDGEMENTS
- Figure
- Reference
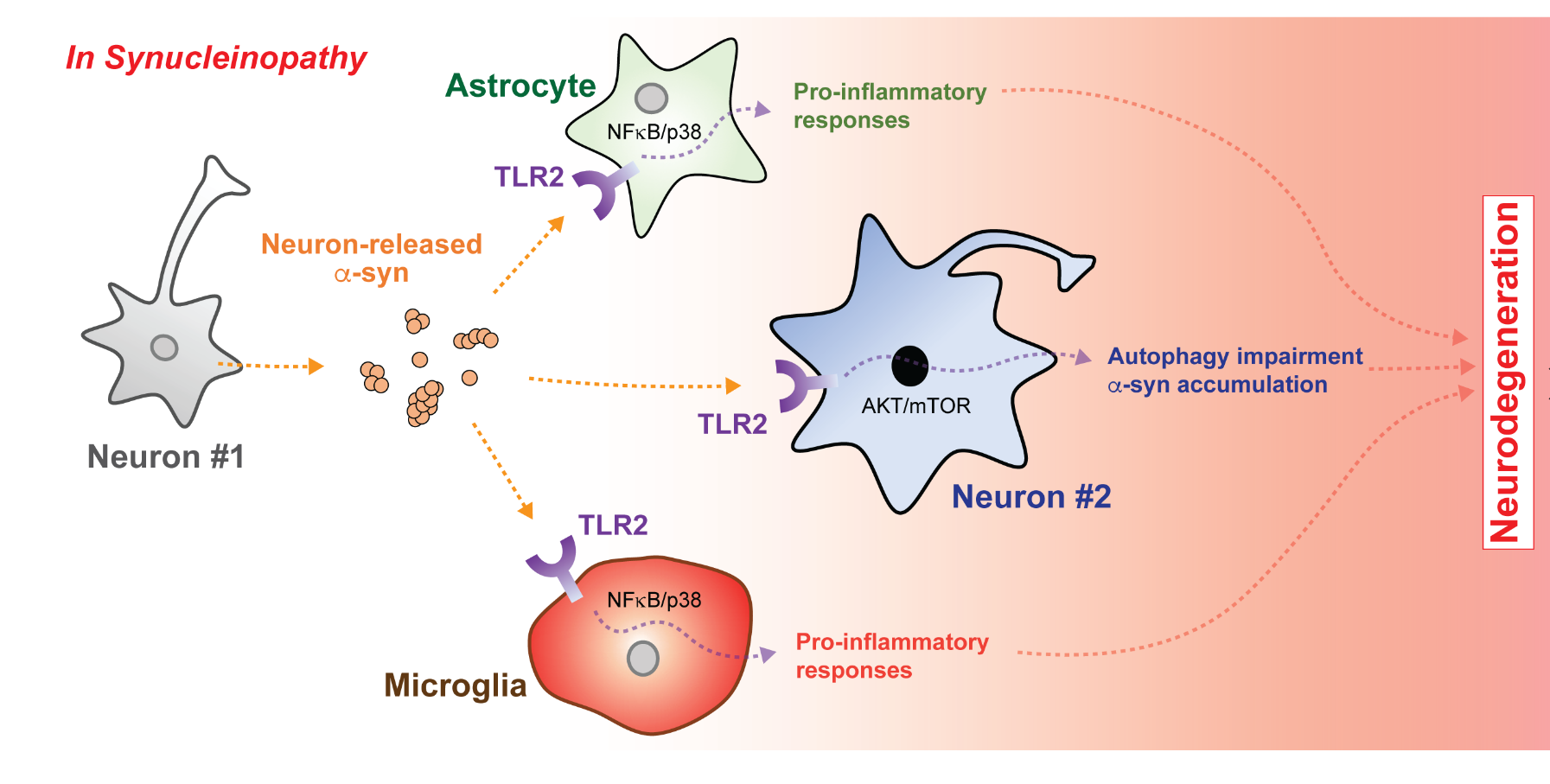
Parkinson’s disease (PD), dementia with Lewy bodies (DLB), multiple system atrophy (MSA), and a subset of Alzheimer’s disease (AD) are collectively referred to as synucleinopathies and characterized by abnormal deposition of α-synuclein (α-syn) in disease-affected brain regions [1, 2]. α-syn is a neuronal protein localized in presynaptic terminals that been shown to be involved in synaptic vesicle trafficking and synaptic plasticity [3, 4]. Under certain conditions, a small amount of α-syn can be released into the extracellular space via unconventional exocytosis [5–7]. Neuron-released α-syn can then stimulate neurotoxic responses in neighboring neurons and glia by interacting with cell surface receptors [8–11]. In microglia, α-syn induces a pro-inflammatory phenotype characterized by enhanced cell migration, oxidative stress, nitric oxide production, and release of cytokines and chemokines [12–15]. The microglial receptors proposed to recognize extracellular α-syn are toll-like receptor 2 (TLR2), TLR4, cluster of differentiation 36 (CD36), macrophage-1 antigen (MAC1), nicotinamide adenine dinucleotide phosphate oxidase 2 (Nox2), and β1-integrin [16–21]. Neurons, on the other hand, may interact with extracellular α-syn via TLR2 and lymphocyte activation gene 3 (LAG3) to modulate autophagy and α-syn transmission, eventually leading to abnormal deposition of α-syn and neuronal death [9, 22, 23]. As such, targeted regulation of these receptors may have beneficial therapeutic effects by alleviating α-syn-mediated toxicity.
TLRs are a family of membrane-bound pattern recognition receptors (PRRs) responsible for an innate immune response. Upon recognizing their specific pathogens, TLRs engage in an assortment of intracellular signaling pathways that can induce the expression of inflammatory genes to regulate the host’s immune response [24]. TLRs are primarily expressed by innate immune cells such as monocytes/macrophages and dendritic cells, but they can also be found on adaptive immune and non-immune cells [25]. In the central nervous system, microglia, astrocytes, neurons, and oligodendrocytes are all known to express certain types of TLRs [26]. Although TLRs have been extensively studied in the innate immune system, recent studies have also suggested a critical role for TLRs in neurodegenerative diseases such as synucleinopathies and AD [27–29]. Here, we will explore α-syn-induced microglial and neuronal TLR2 activation and discuss TLR2 as a potential therapeutic target for synucleinopathies.
PATHOLOGICAL INTERACTION OF MICROGLIAL TLR2 AND EXTRACELLULAR α-SYNUCLEIN
- Go to
- Abstract
- Graphical Abstract
- INTRODUCTION
- PATHOLOGICAL INTERACTION OF MICROGLIAL TLR2 AND EXTRACELLULAR α-SYNUCLEIN
- PATHOLOGICAL INTERACTION OF NEURONAL TLR2 AND EXTRACELLULAR α-SYNUCLEIN
- TLR2 IN CELL-TO-CELL TRANSMISSION
- TLR2 IMMUNOTHERAPY IN SYNUCLEINOPATHY MOUSE MODEL
- CONCLUSIONS
- ACKNOWLEDGEMENTS
- Figure
- Reference
In 2013, we demonstrated the pathological role of microglial TLR2 in synucleinopathies [30]. To investigate the microglial response to neuron-released α-syn, we introduced culture media obtained from differentiated human neuroblastoma SH-SY5Y cells (dSY5Y) overexpressing either wildtype human α-syn (αSCM) or β-galactosidase (LZCM, control conditioned media) to rat primary microglia. Cells treated with αSCM displayed increased amoeboid morphology, cell proliferation, and nitric oxide production, indicating microglial activation. However, αSCM contains not only neuron-released α-syn but also other cellular byproducts. To evaluate whether microglial activation by αSCM depends on the presence of α-syn in the conditioned medium, we serially and selectively eliminated α-syn from αSCM and observed a concomitant decrease in microglial activation. Total depletion of α-syn from the conditioned media completely abolished the microglial response to αSCM. Microglial activation was conversely increased by treatment of α-syn purified from αSCM in a dose-dependent manner.
To gain a comprehensive mechanistic understanding for α-syn-induced microglia activation, we then analyzed the early (6 hr) and late (24 hr) responses in the gene transcriptome of microglia exposed to neuron-released α-syn. Jak-STAT, cytokine-cytokine receptor, leukocyte transendothelial migration, and regulation of actin cytoskeleton pathways were indicated in either the early or late response. On the other hand, TLR2 and downstream signaling proteins myeloid differentiation primary response 88 (Myd88), interleukin-1 receptor-associated kinase (IRAK), nuclear factor kappa-light-chain-enhancer of activated B cells (NFκB), and p38 mitogen-activated protein kinase (MAPK) exhibited sustained activation across time points. Transcriptome analysis of human PD patients also demonstrated an upregulation of TLR2 and downstream signaling cascade components such as CD14, IRAK2, and NFκB.
Given the induction of TLR2 in synucleinopathy, we next examined whether modulation of TLR2 could suppress microglial activation. Primary microglia isolated from Tlr2 knockout (
Neurons release various forms of α-syn, including monomers, oligomers, and high molecular weight aggregates [31–34]. To identify which α-syn conformations interact with TLR2, we sorted neuron-released α-syn by size exclusion chromatography and observed that oligomeric α-syn had the highest TLR2 agonistic activity while low weight oligomers and monomer had little effect. Biophysical analysis further revealed that these oligomers are β-sheet-enriched. In the manner thus described, we concluded that β-sheet-enriched oligomeric forms of neuron-released α-syn can induce microglial neuroinflammation via TLR2 (Fig. 1).
Our findings were supported by the studies that followed. In microglia pretreated with α-syn, administration of a TLR2-specific agonist, but not other TLR agonists, was sufficient to increase cytokine gene expression such as that of IL-6 [35]. Additional studies observed profoundly increased TLR2 expression in the brains of synucleinopathies patients and aged animal models [36, 37]. Our finding that misfolded α-syn activated microglia via MyD88-dependent TLR1/2 signaling was also reinforced [38]. Furthermore, La Vitola et al. showed that α-syn oligomers impaired memory formation in a PD mouse model through a TLR2-dependent process [39]. Qiao et al. additionally demonstrated that TLR2 neutralizing antibody and knockdown could prevent microglial activation by neuron-released α-syn [40]. Similarly, exercise was found to have a neuroprotective effect in a pharmacological animal model of PD by down-regulating TLR2 expression and downstream signaling molecules such as MyD88, tumor necrosis factor receptor-associated factor 6 (TRAF-6), and transforming growth factor β-activated protein kinase 1 (TAK-1) [41, 42]. The literature thus supports that targeting microglia TLR2 might be beneficial to the treatment of synucleinopathies.
PATHOLOGICAL INTERACTION OF NEURONAL TLR2 AND EXTRACELLULAR α-SYNUCLEIN
- Go to
- Abstract
- Graphical Abstract
- INTRODUCTION
- PATHOLOGICAL INTERACTION OF MICROGLIAL TLR2 AND EXTRACELLULAR α-SYNUCLEIN
- PATHOLOGICAL INTERACTION OF NEURONAL TLR2 AND EXTRACELLULAR α-SYNUCLEIN
- TLR2 IN CELL-TO-CELL TRANSMISSION
- TLR2 IMMUNOTHERAPY IN SYNUCLEINOPATHY MOUSE MODEL
- CONCLUSIONS
- ACKNOWLEDGEMENTS
- Figure
- Reference
In a subsequent study, we crossed
To assess this role, we stimulated α-syn-overexpressing neuronal cells with the TLR2-specific agonist, pam3CSK4. Strikingly, stimulation of TLR2 significantly increased the intracellular accumulation of high molecular weight α-syn aggregates, with cytotoxicity. On the other hand, lentiviral vector-mediated knockdown of TLR2 in these cells normalized α-syn accumulation to that of control levels regardless of whether the agonist was introduced. Having observed that TLR2 was associated with α-syn deposition
We next sought to understand the signaling mechanism by which TLR2 affects neuronal α-syn accumulation. Given that α-syn mRNA levels were unaffected by genetic depletion of TLR2 and, in general, TLRs are associated with pathogen clearance, we hypothesized that neuronal α-syn deposition might be associated with cellular protein homeostasis mechanisms such as autophagy [46, 47]. To verify this idea, we monitored proteostasis markers in TLR2-stimulated neurons and demonstrated that activation of TLR2 increased the accumulation of not only high molecular weight α-syn aggregates but also sequestosome 1 (p62/SQSTM1) and puncta formation of microtubule-associated proteins 1A/1B light chain 3B (LC3), which are indicators of autophagy [48]. Treatment of rapamycin, an autophagy inducer, significantly reduced TLR2-mediated α-syn accumulation. Notably, co-treating neurons with TLR2 agonist and autophagy inhibitor bafilomycin A1 did not produce an additive effect on α-syn accumulation, which suggests that the two drugs act along the same pathway.
The mammalian target of rapamycin (mTOR) is a key modulator of autophagy [49]. Given that rapamycin reversed the effects of TLR2 agonist on α-syn pathology, we investigated the role of mTOR signaling in TLR2-mediated neuronal α-syn deposition. Interestingly, activation of TLR2 in α-syn-overexpressing dSY5Y cells increased the inhibitory phosphorylation of mTOR as well as the phosphorylation of AKT, an mTOR negative regulator. As further evidence, stimulation of TLR2 increased the accumulation of p62/SQSTM1 and α-syn in human neural precursor cells overexpressing α-syn. In addition, treatment with an AKT inhibitor or lentiviral knockdown of TLR2 was sufficient to reverse these effects, suggesting that neuronal TLR2 activation by α-syn promotes intracellular α-syn deposition through an AKT/mTOR-dependent inhibition of autophagy. These findings were supported by Dzamko et al, who described the induction of neuronal TLR2 in PD patient brains [50]. Dzamko et al also verified that TLR2 activation disrupts neuronal autophagy and results in α-syn accumulation. As such, neuronal TLR2 might also be a viable therapeutic target for synucleinopathies (Fig. 1).
TLR2 IN CELL-TO-CELL TRANSMISSION
- Go to
- Abstract
- Graphical Abstract
- INTRODUCTION
- PATHOLOGICAL INTERACTION OF MICROGLIAL TLR2 AND EXTRACELLULAR α-SYNUCLEIN
- PATHOLOGICAL INTERACTION OF NEURONAL TLR2 AND EXTRACELLULAR α-SYNUCLEIN
- TLR2 IN CELL-TO-CELL TRANSMISSION
- TLR2 IMMUNOTHERAPY IN SYNUCLEINOPATHY MOUSE MODEL
- CONCLUSIONS
- ACKNOWLEDGEMENTS
- Figure
- Reference
The cell-to-cell transmission of α-syn has also been proposed to play a critical role in synucleinopathy pathogenesis [9, 51, 52]. To assess whether TLR2 is associated with α-syn transmission, we employed a live-cell monitoring system consisting of neuronal donor cells expressing α-syn conjugated to the amino-terminus of the fluorescent protein Venus and neuronal recipient cells expressing α-syn conjugated to the carboxy-terminus of Venus [53, 54]. Intriguingly, induction of TLR2 through overexpression or an agonist increased the cytotoxic neuron-to-neuron transmission of α-syn. However, functional or genetic inhibition of TLR2 suppressed this transmission. These results indicate that TLR2 plays a critical role in the neuron-to-neuron transmission of α-syn aggregates. In addition to neuron-to-neuron transmission, α-syn may also be transferred to neighboring glial cells [8]. Although astrocytes do not express α-syn, we observed significant α-syn accumulation in the astrocytes of both synucleinopathy patients and mouse models [8, 55]. We thus also verified that astrocytic α-syn aggregates originated from neurons in a TLR2-dependent manner [8].
TLR2 IMMUNOTHERAPY IN SYNUCLEINOPATHY MOUSE MODEL
- Go to
- Abstract
- Graphical Abstract
- INTRODUCTION
- PATHOLOGICAL INTERACTION OF MICROGLIAL TLR2 AND EXTRACELLULAR α-SYNUCLEIN
- PATHOLOGICAL INTERACTION OF NEURONAL TLR2 AND EXTRACELLULAR α-SYNUCLEIN
- TLR2 IN CELL-TO-CELL TRANSMISSION
- TLR2 IMMUNOTHERAPY IN SYNUCLEINOPATHY MOUSE MODEL
- CONCLUSIONS
- ACKNOWLEDGEMENTS
- Figure
- Reference
With evidence to support that extracellular α-syn pathologically activates TLR2 in synucleinopathies, we shifted our focus to the therapeutic potential of modulating TLR2 activity [56]. We first verified that TLR2 was present and elevated in the neurons and microglia of synucleinopathy patients and animal models. Then, we administered a TLR2 functional blocking antibody (T2.5) into a synucleinopathy mouse model mimicking the neuropathological and functional aspects of the disease [56]. As expected, blockade of TLR2 successfully diminished α-syn deposition in neurons, especially that of triton-insoluble high molecular weight α-syn aggregates. Treatment with T2.5 also reduced astrogliosis and microgliosis as well as decreased the expression of pro-inflammatory cytokines such as tumor necrosis factors alpha (TNFα) and interleukin 6 (IL-6). Remarkably, T2.5 administration significantly ameliorated neuronal loss in synucleinopathy mice, and this neuroprotective effect was reflected in the improvement of behavioral deficits in the model such as hyperactivity.
Based on these data and our previous findings, we propose three mechanisms by which functional inhibition of TLR2 could alleviate α-syn pathology, inflammation, and neurodegeneration in models of synucleinopathy. First, functional inhibition of TLR2 reduces glial activations and subsequent neuroinflammation [30]. Second, TLR2 blockade relieves the suppression of neuronal autophagy to decrease abnormal deposition of α-syn [43]. Finally, neutralization of TLR2 may inhibit neuron-to-neuron and neuron-to-glia α-syn transmission (Fig. 1).
CONCLUSIONS
- Go to
- Abstract
- Graphical Abstract
- INTRODUCTION
- PATHOLOGICAL INTERACTION OF MICROGLIAL TLR2 AND EXTRACELLULAR α-SYNUCLEIN
- PATHOLOGICAL INTERACTION OF NEURONAL TLR2 AND EXTRACELLULAR α-SYNUCLEIN
- TLR2 IN CELL-TO-CELL TRANSMISSION
- TLR2 IMMUNOTHERAPY IN SYNUCLEINOPATHY MOUSE MODEL
- CONCLUSIONS
- ACKNOWLEDGEMENTS
- Figure
- Reference
TLR2 is involved in the neuropathogenesis of synucleinopathy in
ACKNOWLEDGEMENTS
- Go to
- Abstract
- Graphical Abstract
- INTRODUCTION
- PATHOLOGICAL INTERACTION OF MICROGLIAL TLR2 AND EXTRACELLULAR α-SYNUCLEIN
- PATHOLOGICAL INTERACTION OF NEURONAL TLR2 AND EXTRACELLULAR α-SYNUCLEIN
- TLR2 IN CELL-TO-CELL TRANSMISSION
- TLR2 IMMUNOTHERAPY IN SYNUCLEINOPATHY MOUSE MODEL
- CONCLUSIONS
- ACKNOWLEDGEMENTS
- Figure
- Reference
This research was supported entirely by the Intramural research Program of the National Institutes of Health, National institute on Aging.
Figures
- Go to
- Abstract
- Graphical Abstract
- INTRODUCTION
- PATHOLOGICAL INTERACTION OF MICROGLIAL TLR2 AND EXTRACELLULAR α-SYNUCLEIN
- PATHOLOGICAL INTERACTION OF NEURONAL TLR2 AND EXTRACELLULAR α-SYNUCLEIN
- TLR2 IN CELL-TO-CELL TRANSMISSION
- TLR2 IMMUNOTHERAPY IN SYNUCLEINOPATHY MOUSE MODEL
- CONCLUSIONS
- ACKNOWLEDGEMENTS
- Figure
- Reference
{kind=link}
References
- Go to
- Abstract
- Graphical Abstract
- INTRODUCTION
- PATHOLOGICAL INTERACTION OF MICROGLIAL TLR2 AND EXTRACELLULAR α-SYNUCLEIN
- PATHOLOGICAL INTERACTION OF NEURONAL TLR2 AND EXTRACELLULAR α-SYNUCLEIN
- TLR2 IN CELL-TO-CELL TRANSMISSION
- TLR2 IMMUNOTHERAPY IN SYNUCLEINOPATHY MOUSE MODEL
- CONCLUSIONS
- ACKNOWLEDGEMENTS
- Figure
- Reference
- Spillantini MG, Schmidt ML, Lee VM, Trojanowski JQ, Jakes R, Goedert M (1997) Alpha-synuclein in Lewy bodies. Nature 388:839-840.


- McCann H, Stevens CH, Cartwright H, Halliday GM (2014) α-Synucleinopathy phenotypes. Parkinsonism Relat Disord 20 Suppl 1:S62-S67.
- Masliah E, Rockenstein E, Veinbergs I, Mallory M, Hashimoto M, Takeda A, Sagara Y, Sisk A, Mucke L (2000) Dopaminergic loss and inclusion body formation in alpha-synuclein mice: implications for neurodegenerative disorders. Science 287:1265-1269.
- Lashuel HA, Overk CR, Oueslati A, Masliah E (2013) The many faces of α-synuclein: from structure and toxicity to therapeutic target. Nat Rev Neurosci 14:38-48.

- Lee HJ, Patel S, Lee SJ (2005) Intravesicular localization and exocytosis of alpha-synuclein and its aggregates. J Neurosci 25:6016-6024.
- Jang A, Lee HJ, Suk JE, Jung JW, Kim KP, Lee SJ (2010) Nonclassical exocytosis of alpha-synuclein is sensitive to folding states and promoted under stress conditions. J Neurochem 113:1263-1274.
- Lee HJ, Cho ED, Lee KW, Kim JH, Cho SG, Lee SJ (2013) Autophagic failure promotes the exocytosis and intercellular transfer of α-synuclein. Exp Mol Med 45:e22.
- Lee HJ, Suk JE, Patrick C, Bae EJ, Cho JH, Rho S, Hwang D, Masliah E, Lee SJ (2010) Direct transfer of alpha-synuclein from neuron to astroglia causes inflammatory responses in synucleinopathies. J Biol Chem 285:9262-9272.
- Desplats P, Lee HJ, Bae EJ, Patrick C, Rockenstein E, Crews L, Spencer B, Masliah E, Lee SJ (2009) Inclusion formation and neuronal cell death through neuron-to-neuron transmission of alpha-synuclein. Proc Natl Acad Sci U S A 106:13010-13015.
- Chung CY, Khurana V, Auluck PK, Tardiff DF, Mazzulli JR, Soldner F, Baru V, Lou Y, Freyzon Y, Cho S, Mungenast AE, Muffat J, Mitalipova M, Pluth MD, Jui NT, Schüle B, Lippard SJ, Tsai LH, Krainc D, Buchwald SL, Jaenisch R, Lindquist S (2013) Identification and rescue of α-synuclein toxicity in Parkinson patient-derived neurons. Science 342:983-987.
- Theodore S, Cao S, McLean PJ, Standaert DG (2008) Targeted overexpression of human alpha-synuclein triggers microglial activation and an adaptive immune response in a mouse model of Parkinson disease. J Neuropathol Exp Neurol 67:1149-1158.
- Long-Smith CM, Sullivan AM, Nolan YM (2009) The influence of microglia on the pathogenesis of Parkinson's disease. Prog Neurobiol 89:277-287.
- Shimohama S, Tanino H, Kawakami N, Okamura N, Kodama H, Yamaguchi T, Hayakawa T, Nunomura A, Chiba S, Perry G, Smith MA, Fujimoto S (2000) Activation of NADPH oxidase in Alzheimer's disease brains. Biochem Biophys Res Commun 273:5-9.
- Giasson BI, Duda JE, Murray IV, Chen Q, Souza JM, Hurtig HI, Ischiropoulos H, Trojanowski JQ, Lee VM (2000) Oxidative damage linked to neurodegeneration by selective alphasynuclein nitration in synucleinopathy lesions. Science 290:985-989.
- Wang S, Chu CH, Stewart T, Ginghina C, Wang Y, Nie H, Guo M, Wilson B, Hong JS, Zhang J (2015) α-Synuclein, a chemoattractant, directs microglial migration via H2O2- dependent Lyn phosphorylation. Proc Natl Acad Sci U S A 112:E1926-E1935.
- Fellner L, Irschick R, Schanda K, Reindl M, Klimaschewski L, Poewe W, Wenning GK, Stefanova N (2013) Toll-like receptor 4 is required for α-synuclein dependent activation of microglia and astroglia. Glia 61:349-360.
- Stewart CR, Stuart LM, Wilkinson K, van Gils JM, Deng J, Halle A, Rayner KJ, Boyer L, Zhong R, Frazier WA, Lacy-Hulbert A, El Khoury J, Golenbock DT, Moore KJ (2010) CD36 ligands promote sterile inflammation through assembly of a Toll-like receptor 4 and 6 heterodimer. Nat Immunol 11:155-161.
- Udan ML, Ajit D, Crouse NR, Nichols MR (2008) Toll-like receptors 2 and 4 mediate Abeta(1-42) activation of the innate immune response in a human monocytic cell line. J Neurochem 104:524-533.
- Zhang W, Dallas S, Zhang D, Guo JP, Pang H, Wilson B, Miller DS, Chen B, Zhang W, McGeer PL, Hong JS, Zhang J (2007) Microglial PHOX and Mac-1 are essential to the enhanced dopaminergic neurodegeneration elicited by A30P and A53T mutant alpha-synuclein. Glia 55:1178-1188.
- Jiang T, Hoekstra J, Heng X, Kang W, Ding J, Liu J, Chen S, Zhang J (2015) P2X7 receptor is critical in α-synuclein-- mediated microglial NADPH oxidase activation. Neurobiol Aging 36:2304-2318.
- Kim C, Cho ED, Kim HK, You S, Lee HJ, Hwang D, Lee SJ (2014) β1-integrin-dependent migration of microglia in response to neuron-released α-synuclein. Exp Mol Med 46:e91.
- Liu HY, Chen CY, Hsueh YP (2014) Innate immune responses regulate morphogenesis and degeneration: roles of Toll-like receptors and Sarm1 in neurons. Neurosci Bull 30:645-654.
- Mao X, Ou MT, Karuppagounder SS, Kam TI, Yin X, Xiong Y, Ge P, Umanah GE, Brahmachari S, Shin JH, Kang HC, Zhang J, Xu J, Chen R, Park H, Andrabi SA, Kang SU, Gonçalves RA, Liang Y, Zhang S, Qi C, Lam S, Keiler JA, Tyson J, Kim D, Panicker N, Yun SP, Workman CJ, Vignali DA, Dawson VL, Ko HS, Dawson TM (2016) Pathological α-synuclein transmission initiated by binding lymphocyte-activation gene 3. Science 353:aah3374.
- Medzhitov R (2001) Toll-like receptors and innate immunity. Nat Rev Immunol 1:135-145.
- Liu Y, Yin H, Zhao M, Lu Q TL (2014) TLR2 and TLR4 in autoimmune diseases: a comprehensive review. Clin Rev Allergy Immunol 47:136-147.
- Lee H, Lee S, Cho IH, Lee SJ (2013) Toll-like receptors: sensor molecules for detecting damage to the nervous system. Curr Protein Pept Sci 14:33-42.
- Li X, Xue L, Sun J, Sun Y, Xie A (2017) Single nucleotide polymorphisms in the toll-like receptor 2 (TLR2) gene are associated with sporadic Parkinson's disease in the North-eastern Han Chinese population. Neurosci Lett 656:72-76.
- Caplan IF, Maguire-Zeiss KA (2018) Toll-like receptor 2 signaling and current approaches for therapeutic modulation in synucleinopathies. Front Pharmacol 9:417.
- Gąsiorowski K, Brokos B, Echeverria V, Barreto GE, Leszek J (2018) RAGE-TLR crosstalk sustains chronic inflammation in neurodegeneration. Mol Neurobiol 55:1463-1476.
- Kim C, Ho DH, Suk JE, You S, Michael S, Kang J, Joong Lee S, Masliah E, Hwang D, Lee HJ, Lee SJ (2013) Neuron-released oligomeric α-synuclein is an endogenous agonist of TLR2 for paracrine activation of microglia. Nat Commun 4:1562.
- Park JY, Paik SR, Jou I, Park SM (2008) Microglial phagocytosis is enhanced by monomeric alpha-synuclein, not aggregated alpha-synuclein: implications for Parkinson's disease. Glia 56:1215-1223.
- Reynolds AD, Glanzer JG, Kadiu I, Ricardo-Dukelow M, Chaudhuri A, Ciborowski P, Cerny R, Gelman B, Thomas MP, Mosley RL, Gendelman HE (2008) Nitrated alpha-synucleinactivated microglial profiling for Parkinson's disease. J Neurochem 104:1504-1525.
- Su X, Maguire-Zeiss KA, Giuliano R, Prifti L, Venkatesh K, Federoff HJ (2008) Synuclein activates microglia in a model of Parkinson's disease. Neurobiol Aging 29:1690-1701.
- Zhang W, Wang T, Pei Z, Miller DS, Wu X, Block ML, Wilson B, Zhang W, Zhou Y, Hong JS, Zhang J (2005) Aggregated alphasynuclein activates microglia: a process leading to disease progression in Parkinson's disease. FASEB J 19:533-542.
- Roodveldt C, Labrador-Garrido A, Gonzalez-Rey E, Lachaud CC, Guilliams T, Fernandez-Montesinos R, Benitez-Rondan A, Robledo G, Hmadcha A, Delgado M, Dobson CM, Pozo D (2013) Preconditioning of microglia by α-synuclein strongly affects the response induced by toll-like receptor (TLR) stimulation. PLoS One 8:e79160.
- Doorn KJ, Moors T, Drukarch B, van de Berg WDj, Lucassen PJ, van Dam AM (2014) Microglial phenotypes and tolllike receptor 2 in the substantia nigra and hippocampus of incidental Lewy body disease cases and Parkinson's disease patients. Acta Neuropathol Commun 2:90.
- Drouin-Ouellet J, St-Amour I, Saint-Pierre M, Lamontagne- Proulx J, Kriz J, Barker RA, Cicchetti F (2014) Toll-like receptor expression in the blood and brain of patients and a mouse model of Parkinson's disease. Int J Neuropsychopharmacol 18:pyu103.
- Daniele SG, Béraud D, Davenport C, Cheng K, Yin H, Maguire-Zeiss KA (2015) Activation of MyD88-dependent TLR1/2 signaling by misfolded α-synuclein, a protein linked to neurodegenerative disorders. Sci Signal 8:ra45.
- La Vitola P, Balducci C, Cerovic M, Santamaria G, Brandi E, Grandi F, Caldinelli L, Colombo L, Morgese MG, Trabace L, Pollegioni L, Albani D, Forloni G (2018) Alpha-synuclein oligomers impair memory through glial cell activation and via Toll-like receptor 2. Brain Behav Immun 69:591-602.
- Qiao H, Zhang Q, Yuan H, Li Y, Wang D, Wang R, He X (2015) Elevated neuronal α-synuclein promotes microglia activation after spinal cord ischemic/reperfused injury. Neuroreport 26:656-661.
- Jang Y, Koo JH, Kwon I, Kang EB, Um HS, Soya H, Lee Y, Cho JY (2017) Neuroprotective effects of endurance exercise against neuroinflammation in MPTP-induced Parkinson's disease mice. Brain Res 1655:186-193.
- Koo JH, Jang YC, Hwang DJ, Um HS, Lee NH, Jung JH, Cho JY (2017) Treadmill exercise produces neuroprotective effects in a murine model of Parkinson's disease by regulating the TLR2/MyD88/NF-κB signaling pathway. Neuroscience 356:102-113.
- Kim C, Rockenstein E, Spencer B, Kim HK, Adame A, Trejo M, Stafa K, Lee HJ, Lee SJ, Masliah E (2015) Antagonizing neuronal toll-like receptor 2 prevents synucleinopathy by activating autophagy. Cell Rep 13:771-782.
- Chen CY, Lin CW, Chang CY, Jiang ST, Hsueh YP (2011) Sarm1, a negative regulator of innate immunity, interacts with syndecan-2 and regulates neuronal morphology. J Cell Biol 193:769-784.
- Okun E, Griffioen KJ, Mattson MP (2011) Toll-like receptor signaling in neural plasticity and disease. Trends Neurosci 34:269-281.
- Schmitz F, Heit A, Dreher S, Eisenächer K, Mages J, Haas T, Krug A, Janssen KP, Kirschning CJ, Wagner H (2008) Mammalian target of rapamycin (mTOR) orchestrates the defense program of innate immune cells. Eur J Immunol 38:2981-2992.
- Klionsky DJ (2007) Autophagy: from phenomenology to molecular understanding in less than a decade. Nat Rev Mol Cell Biol 8:931-937.
- Sahani MH, Itakura E2, Mizushima N (2014) Expression of the autophagy substrate SQSTM1/p62 is restored during prolonged starvation depending on transcriptional upregulation and autophagy-derived amino acids. Autophagy 10:431-441.
- Janku F, McConkey DJ, Hong DS, Kurzrock R (2011) Autophagy as a target for anticancer therapy. Nat Rev Clin Oncol 8:528-539.
- Dzamko N, Gysbers A, Perera G, Bahar A, Shankar A, Gao J, Fu Y, Halliday GM (2017) Toll-like receptor 2 is increased in neurons in Parkinson's disease brain and may contribute to alpha-synuclein pathology. Acta Neuropathol 133:303-319.
- Danzer KM, Kranich LR, Ruf WP, Cagsal-Getkin O, Winslow AR, Zhu L, Vanderburg CR, McLean PJ (2012) Exosomal cell-to-cell transmission of alpha synuclein oligomers. Mol Neurodegener 7:42.
- Hansen C, Angot E, Bergström AL, Steiner JA, Pieri L, Paul G, Outeiro TF, Melki R, Kallunki P, Fog K, Li JY, Brundin P (2011) α-Synuclein propagates from mouse brain to grafted dopaminergic neurons and seeds aggregation in cultured human cells. J Clin Invest 121:715-725.
- Bae EJ, Yang NY, Song M, Lee CS, Lee JS, Jung BC, Lee HJ, Kim S, Masliah E, Sardi SP, Lee SJ (2014) Glucocerebrosidase depletion enhances cell-to-cell transmission of α-synuclein. Nat Commun 5:4755.
- Kerppola TK (2006) Design and implementation of bimolecular fluorescence complementation (BiFC) assays for the visualization of protein interactions in living cells. Nat Protoc 1:1278-1286.
- Halliday GM, Stevens CH (2011) Glia: initiators and progressors of pathology in Parkinson's disease. Mov Disord 26:6-17.
- Kim C, Spencer B, Rockenstein E, Yamakado H, Mante M, Adame A, Fields JA, Masliah D, Iba M, Lee HJ, Rissman RA, Lee SJ, Masliah E (2018) Immunotherapy targeting toll-like receptor 2 alleviates neurodegeneration in models of synucleinopathy by modulating α-synuclein transmission and neuroinflammation. Mol Neurodegener 13:43.