Articles

Article Tools
 View Full Text View Full Text |
 Abstract Abstract |
 Article as PDF Article as PDF |
 Print this Article Print this Article |
 Pubmed Pubmed |
 PMC PMC |
 PubReader PubReader |
 Export to Citation Export to Citation |
 Email Alerts Email Alerts |
 Open Access Open Access |
Share this article on :



Stats or Metrics
Article
Original Article
Exp Neurobiol 2019; 28(4): 458-473
Published online August 31, 2019
https://doi.org/10.5607/en.2019.28.4.458
© The Korean Society for Brain and Neural Sciences
Increased CD68/TGFβ Co-expressing Microglia/ Macrophages after Transient Middle Cerebral Artery Occlusion in Rhesus Monkeys
Hyeon-Gu Yeo1,2†, Jung Joo Hong1†, Youngjeon Lee1,2†, Kyung Sik Yi3, Chang-Yeop Jeon1, Junghyung Park1, Jinyoung Won1, Jincheol Seo1,4, Yu-Jin Ahn1,2, Keonwoo Kim1,5, Seung Ho Baek1, Eun-Ha Hwang1,6, Green Kim1,6, Yeung Bae Jin1, Kang-Jin Jeong1, Bon-Sang Koo1, Philyong Kang7, Kyung Seob Lim7, Sun-Uk Kim2,7, Jae-Won Huh1,2, Young-Hyun Kim1,2, Yeonghoon Son8, Ji-Su Kim8, Chi-Hoon Choi3, Sang-Hoon Cha3* and Sang-Rae Lee1,2*
1National Primate Research Center, Korea Research Institute of Bioscience and Biotechnology (KRIBB), Cheongju 28116, 2Department of Functional Genomics, KRIBB School of Bioscience, Korea University of Science and Technology, Daejeon 34113, 3Department of Radiology, Chungbuk National University Hospital, Cheongju 28644, 4School of Life Sciences, BK21 Plus KNU Creative BioResearch Group, Kyungpook National University, Daegu 41566, 5Department of Physical Therapy, Graduate School of Inje University, Gimhae 50834, 6Laboratory Animal Medicine, College of Veterinary Medicine, Chonnam National University, Gwangju 61186, 7Futuristic Animal Resource & Research Center, KRIBB, Cheongju 28116, 8Primate Resource Center, KRIBB, Jeongeup 56216, Korea
Correspondence to: *To whom correspondence should be addressed.
Sang-Rae Lee, TEL: 82-43-240-6316, FAX: 82-43-240-6309, e-mail: srlee@kribb.re.kr
Sang-Hoon Cha, TEL: 82-43-269-6473, FAX: 82-43-269-6479, e-mail: shcha@chungbuk.ac.kr
†These authors contributed equally to work.
This is an Open Access article distributed under the terms of the Creative Commons Attribution Non-Commercial License(http://creativecommons.org/licenses/by-nc/4.0) which permits unrestricted non-commercial use, distribution, andreproduction in any medium, provided the original work is properly cited.
Abstract
The function of microglia/macrophages after ischemic stroke is poorly understood. This study examines the role of microglia/macrophages in the focal infarct area after transient middle cerebral artery occlusion (MCAO) in rhesus monkeys. We measured infarct volume and neurological function by magnetic resonance imaging (MRI) and non-human primate stroke scale (NHPSS), respectively, to assess temporal changes following MCAO. Activated phagocytic microglia/macrophages were examined by immunohistochemistry in post-mortem brains (n=6 MCAO, n=2 controls) at 3 and 24 hours (acute stage), 2 and 4 weeks (subacute stage), and 4, and 20 months (chronic stage) following MCAO. We found that the infarct volume progressively decreased between 1 and 4 weeks following MCAO, in parallel with the neurological recovery. Greater presence of cluster of differentiation 68 (CD68)-expressing microglia/macrophages was detected in the infarct lesion in the subacute and chronic stage, compared to the acute stage. Surprisingly, 98~99% of transforming growth factor beta (TGFβ) was found colocalized with CD68-expressing cells. CD68-expressing microglia/macrophages, rather than CD206+ cells, may exert anti-inflammatory effects by secreting TGFβ after the subacute stage of ischemic stroke. CD68+ microglia/macrophages can therefore be used as a potential therapeutic target.
Graphical Abstract
Keywords: Inflammation, Microglia, Stroke, Macaca mulatta, Transforming growth factor beta
INTRODUCTION
Stroke is the second leading cause of mortality globally, causing 6.7 million deaths each year [1, 2]. Treatments that can reperfuse the ischemic brain regions as early as possible after stroke onset are critical for the prevention of brain-cell death. As an example, recombinant tissue plasminogen activator is a clot-dissolving agent approved by the Food and Drug Administration despite its limited utility [3]. Another therapeutic option for acute ischemic stroke caused by an abrupt obstruction of a large vessel is endovascular recanalization, which involves the use of various intra-arterial devices to mechanically re-open the steno-occlusive lumen. However, few treatments can be effectively applied in the later stage of ischemic stroke. Moreover, little is known about the therapeutic potential of endogenous recovery mechanisms, warranting further study.
The ischemic brain lesions, visualized as abnormalities on diffusion magnetic resonance imaging (MRI), are complex pathological structures. A patient’s prognosis may depend on the activation of inflammatory cells in the infarcted brain region following the acute ischemic stroke [4]. Many studies using mouse models of ischemic stroke have identified the cluster of differentiation 68 (CD68) as a general marker of activated phagocytic microglia/ macrophages [5], which are the main inflammatory cells in the central nervous system [6]. However, very few studies have investigated the anti-inflammatory activities of CD68-expressing cells during the course of ischemic stroke in rodent models. High numbers of CD68-expressing microglia/macrophages were detected in the infarct core and the border zone at 24 hours and 7 days after ischemic stroke, respectively [7, 8]. In the chronic stage, cells in the ischemic penumbra express transforming growth factor beta (TGFβ), a cytokine involved in neuroprotective functions such as scar formation, inhibition of apoptosis and excitotoxicity, and promotion of angiogenesis and neurogenesis.
Additionally, microglia, particularly the M2 type, secrete cyto-kines with anti-inflammatory functions [9]. The representative M2 microglial marker, CD206, has been identified in central nervous system disorders such as Alzheimer’s disease [10], Parkinson’s disease [11], and acquired immunodeficiency syndrome [12]. In fact, the number of CD206-positive (CD206+) cells in the cortex and striatum significantly increases 3~7 days after middle cerebral artery occlusion (MCAO) [13]. The expression of TGFβ, inter-leukin-10 (IL10), and CD206 mRNA also increases at 24 hours, reaching peak levels 4~6 days after ischemic stroke [14]. The anti-inflammatory activities of CD206+ cells generally decrease around 7 days after ischemic stroke [15]. Elucidation of the progressive alterations in immune cells during the clinical course of stroke are essential for the development of effective therapies.
Histopathological investigations of microglial activation in patients who underwent endovascular recanalization treatment after stroke onset are challenging. To overcome the limitations of clinical studies, we used a non-human primate transient MCAO model established in our previous study [15], which exhibits physiological, anatomical, behavioral, imaging, and genetic properties similar to those observed in humans stroke patients.
MATERIALS AND METHODS
Experimental animals
Eleven rhesus monkeys (Macaca mulatta) were analysed, with brain immunohistological analysis performed in 8 animals postmortem and the remaining 3 monkeys surviving the study to date. Seven animals (R326, R330, R328, R315, R305, R306, and R313) from our previously published experiments [15] and additional two monkeys (R324 and R307) were included in this study. Two age-matched healthy monkeys (R310 and R322) were used as controls (Table 1). The acute (<7 days), subacute (7~30 days), and chronic stages (>30 days) of ischemic stroke were defined based on clinical observations of symptoms and recovery [16]. All experimental animals were considered “young” according to the age classification standards for humans and macaques [17, 18]. The rhesus monkeys (obtained from Gaoyao Kangda Laboratory Animals Science & Technology Co., Ltd., Gaoyao, China), were housed indoors in individual cages at the National Primate Research Center of the Republic of Korea and provided with commercial monkey chow (Harlan, USA) along with a daily supplement of various fruits and water
Ischemic stroke model and MRI
Ischemic stroke was induced using our previously reported endovascular MCAO technique [15]. Immediately after MCAO induction, the animal was transported to the MRI suite while maintaining the endovascular MCAO and scanned using 3.0 T MRI (Achieva 3.0 T, Philips Medical Systems, Best, Netherlands). To identify the infarct core in acute stage and diffusion-perfusion mismatch, diffusion weighted imaging (DWI) was performed at 30 minutes after the MCAO procedure, and MRI acquisition was repeated every 15 minutes thereafter. After the in-bore reperfusion and repeated MRI, postoperative veterinary care was provided to each animal. Infarct volume was measured by follow-up fluid-attenuated inversion recovery (FLAIR) MRI imaging performed once a week until 4 weeks post-infarct. Neuroimaging protocols used for the procedures performed on the day of stroke induction and the follow-up MRI studies were the same as those previously described.
Infarct volume measurement
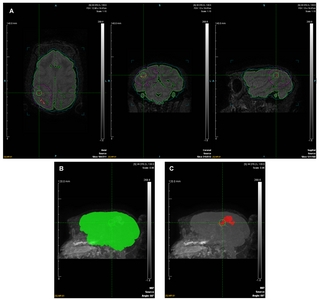
Whole-head FLAIR images were cropped to box dimensions (x=79.50 mm, y=129.75 mm, and z=78.96 mm) to facilitate subsequent analysis (Fig. 1A). Hyper-intense regions were identified, and regions of interest (ROIs) were segmented using the Inveon Research Workplace (IRW; Siemens Medical Solutions, USA) software. Representative ROIs and 3D maximum intensity projection (MIP) views (66o, 0.96 scale; Fig. 1B and 1C), as well as raw data (Table 2), are shown. The following formula was used to calculate the infarct percentage:
To retain experimental objectivity, ROIs were automatically determined using equivalent thresholds: whole brain (38.0/251.0), infarct area (81.0/251.0), and window level (−1.6/268.6). To avoid areas affected by noise, ROI templates were applied manually, excluding the contralateral hemisphere, cerebrospinal fluid, peri-ventricle, and edge signals. ROIs based on representative FLAIR images focused on slice 315/512 coronal section (axial field of view (FOV)=12.98×19.47 cm, coronal FOV=13×19.47 cm, sagittal FOV=13×19.47 cm, and scale=1.15; Fig. 1A). Four ROIs were selected, representing the whole brain (green; RGB, 0.255.0), infarct lesion (red; RGB, 255.0.0), brain template (light blue; RGB, 0.204.204), and infarction template (purple; RGB, 255.0.255). Mean, standard deviation (SD), minimum, and maximum were automatically calculated using IRW for the brain regions, excluding the template region (Table 2).
Behavioral assessment
Neurological scoring was conducted using the validated Non-human Primate Stroke Scale (NHPSS) [21, 22]. NHPSS comprises ratings of state of consciousness, defense reaction, grasp reflex, extremity movement, gait, circling, bradykinesia, balance, neglect, visual field cut/hemianopsia, and facial weakness. Many of the NHPSS ratings reflect the National Institutes of Health Stroke Scoring system in humans. Total scores range from 0 to 41, with 0 corresponding to normal behavior and 41 to severe bilateral neurological impairment. Five monkeys which survived (R328 and R315) or still survive (R305, R306, and R313) beyond 4 weeks into the later chronic stage were evaluated at 1, 3, 7, 9, 13, 16, 20, 23, 27, and 30 days after reperfusion.
Tissue fixation and slice preparation
The post-mortem brains obtained within our previous study [15] (n=4, subacute and chronic groups), comprising an additional acute (n=2) and control groups (n=2), were evaluated. As described in our histological study [20], the brains of experimental animals were fixed with 10% neutral buffered formaldehyde (NBF). The brains were subsequently harvested and post-fixed for 7 days in 10% NBF. The whole brains were sliced in the coronal plane; 4-mm-thick slices were obtained and embedded in a rhesus monkey brain matrix (Ted Pella Inc., Redding, CA, US). Coronal slices (15 to 17) were acquired and numbered from rostral to caudal. Subsequently, the tissues were embedded in paraffin using a routine procedure.
Immunohistochemistry analysis
For immunohistochemistry, 5-μm sections were de-paraffinized, and heat-induced antigen retrieval was performed at 110°C in citrate buffer (pH 6.0; Vector Laboratories, Peterborough, UK). The sections were blocked for 45 minutes with 2% normal goat serum and then incubated for 12 hours at 4°C with the following primary antibodies: polyclonal rabbit anti-ionized calcium binding adaptor molecule 1 (Iba1; 1:500; Abcam, Cambridge, UK), monoclonal mouse anti-CD68 (1:500; Abcam), polyclonal rabbit anti-CD206 (1:100; Abcam), monoclonal rabbit anti-inducible nitric oxide synthase (iNOS; 1:500; Cell Signaling, Danvers, US), polyclonal rabbit anti-tumor necrosis factor-alpha (TNFα; 1:100; Antibodies, Atlanta, US), polyclonal rabbit anti-IL6 (1:100; Antibodies), polyclonal rabbit anti-TGFβ (1:500; Antibodies), polyclonal rabbit anti-IL10 (1:100; Abcam), monoclonal rabbit anti-CD13 (1:500; LSBio, Seattle, US), monoclonal mouse anti-Ki67 (1:100; Abcam), and monoclonal rabbit anti-Ki67 (1:100; Cell Marque, Darmstadt, Germany). For immunofluorescence analysis, sections were incubated at 25°C with CFTM488/594A-labeled goat anti-rabbit and anti-mouse secondary antibody (Biotium, Hayward, CA, US) in darkness for 1 hour. Next, sections were mounted with 4, 6-diamidino-2-phenylindole (DAPI) Vectashield Hard Set Mounting Medium (Vector Laboratories) for nuclei counterstaining, and images were acquired using an Olympus BX51 microscope.
Histological data analysis and quantification
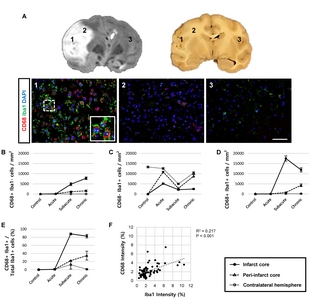
Twenty-five ROIs (400× magnification, FOV=69×52 μm) were selected in the infarct core, peri-infarct core, and the contralateral hemisphere. Representative images were classified into three areas (1, infarct core; 2, peri-infarct; 3, contralateral hemisphere) based on the histological study [23]. Signal intensity was quantified using Image J histogram with inverted 8-bit. Threshold values were as follows: Iba1, 16~23; CD68, 20~25; CD206, 15~20; and TGFβ, 15~20. To remove outliers, both radius (4) and threshold (10) were identically applied. The microglial cells, phagocytic cells, inflammatory cytokine-secreting cells, M1/M2 type microglia/macrophages, and proliferating cells were counted using Image J cell counter plugin.
Statistical analysis
Statistical analysis was performed using the Statistical Package for the Social Sciences for Windows Version, 18.0 (SPSS Inc., Chicago, IL, USA). Wilcoxon signed-rank test was used for pairwise analyses when the Friedman p-value was <0.05. Data were presented as mean±SEM. Linear regression was used for two-group comparisons.
RESULTS
Changes in the infarct volume and behavioral assessments
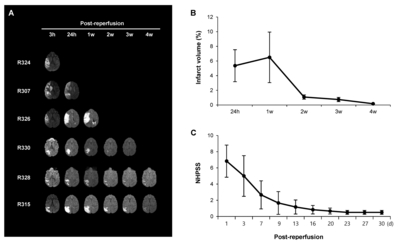
We performed MRI once a week until 4 weeks after ischemic stroke to confirm the presence of the infarct lesion and calculate the infarct volume based on FLAIR images. Representative DWI and FLAIR images are shown in Fig. 2A. The acquired MR data were used to calculate changes in the infarct volume from 24 hours to 4 weeks after reperfusion (Fig. 2B). Interestingly, the infarct volumes exhibited significant reductions between 1 and 2 weeks post-reperfusion compared to the early time points.
Neurobehavioral assessments were conducted using the NHPSS at 1~30 days after stroke (Fig. 2C). Animals subjected to MCAO/ reperfusion showed an initial peak in score at 1 day, followed by a gradual reduction between 9 and 30 days. Since NHPSS total score of 0 corresponds to normal behavior, these results suggest that the neurological impairment in animals recovers almost to the baseline between 9 and 30 days after MCAO. The pattern of change in behavioral outcomes is similar to that of infarct volumes measured by MRI, suggesting that the infarct size reduction may be beneficial.
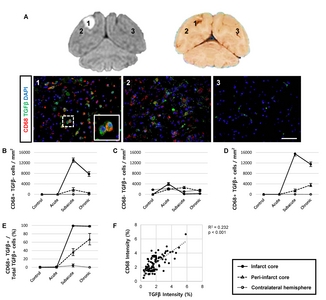
CD68 expression in Iba1+ microglial cells in the ipsilateral hemisphere
First, we analysed the function of activated microglial cells using Iba1 and CD68 as markers for microglia and phagocytic microglia/macrophages, respectively. We detected significantly more cells that were positive for both CD68 and Iba1 in MCAO animals in the subacute and chronic stages, compared to the acute stage, as well as to controls (Fig. 3). Moreover, CD68+ cells were frequently surrounded by cells expressing Iba1 (Fig. 3A). Most of the Iba1+ microglial cells (82~88%) were positive for CD68 in the infarct core during the subacute and chronic stages (Fig. 3D and 3E). Linear regression analysis revealed that the staining intensities of CD68 and Iba1 were significantly correlated in the subacute and chronic stages (Fig. 3F), but not in the acute stage (data not shown). Cells positive for only one of the markers (CD68+/Iba1− or CD68−/ Iba1+ cells) were also found (Fig. 3B and 3C).
TGFβ expression in activated CD68+ cells in the ipsilateral hemisphere
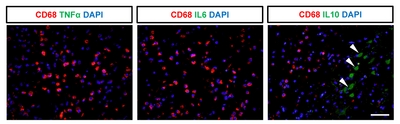
We next evaluated the expression of cytokines to determine whether CD68-expressing cells exhibit inflammatory activity. Immunohistochemistry was performed for pro-inflammatory cytokines, including IL6 and TNFα, and for anti-inflammatory cytokines, including IL10 and TGFβ (Fig. 4 and 5). We detected a significantly higher number of cells positive for both CD68 and TGFβ in the subacute and chronic stages, compared to the acute stage or control animals (Fig. 4D). Indeed, most of the TGFβ+ cells (98~99%) co-expressed CD68 in the chronic stage (Fig. 4E) with significant (p<0.001) linear regression of the intensity observed in the ipsilateral hemisphere of animals in subacute and chronic groups (Fig. 4F), suggesting their anti-inflammatory function. In contrast, cells expressing pro-inflammatory cytokines, such as IL6 and TNFα, were not significantly correlated with CD68-expressing cells (Fig. 5). Interestingly, cells expressing IL10 did not co-express CD68 but were abundantly detected, suggesting that other brain cells, such as astrocytes or effector T cells, may show anti-inflammatory functions.
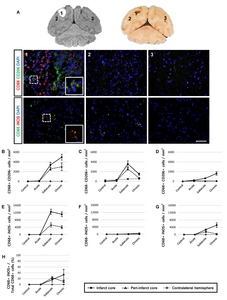
Characterization of M1 and M2 polarization of CD68+ cells
Next, we performed additional histological analyses to determine whether CD68-expressing cells represented the M1 or M2 microglial phenotype. According to the immunological studies [24, 25], a panel of markers for identifying M1 (inducible nitric oxide synthase: iNOS) and M2 (CD206) macrophage phenotypes was used (Fig. 6). Only a few CD68/CD206 double-positive cells were detected in the infarct core (Fig. 6D). Notably, 10~34% of iNOS was co-expressed in the CD68-expressing cells in the ipsilateral hemisphere from the subacute and chronic groups. Single positive cells (CD68+/CD206−, CD68−/CD206+, CD68+/iNOS−, and CD68−/ iNOS+ cells) were also found (Fig. 6B, 6C, 6E and 6F).
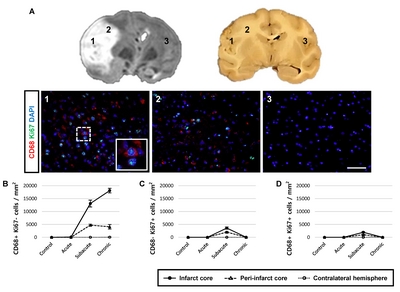
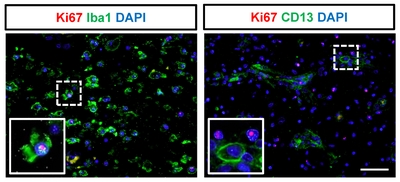
Cell proliferation in the ipsilateral hemisphere after stroke
Lastly, we examined Ki67 expression, a marker of proliferation, in CD68-expressing cells, Iba1+ microglial cells, and CD13+ pericytes (Fig. 7 and 8) after ischemic stroke. Although most of the proliferating cells were negative for CD68 (Fig. 7D) and CD13 (Fig. 8) after ischemic stroke, a few CD68+ proliferating cells were present in the subacute stage. We also observed cells positive for only one of the markers (CD68+/Ki67− cells, Fig. 7B; CD68−/Ki67+ cells, Fig. 7C). Some of the Iba1+ microglial cells exhibited Ki67+ nuclei (Fig. 8).
DISCUSSION
In our previous study, we established an experimental non-human primate stroke model to examine the reversibility of diffusion abnormalities [15]. Clinically, it is difficult to study the alterations in cellular pathology that occur in parallel with diffusion changes in the human brain following the recanalization of the occluded vessel. Therefore, the non-human primate MCAO model can help improve our understanding of how reversible changes in diffusion interact with cellular pathology. Using MRI analysis, Liu et al. identified a true permanent reversal in a non-human primate ischemic stroke model induced by endovascular techniques [26]. However, most experimental ischemic stroke studies have used rodent MCAO models [27–31].
The results of the present study suggest that CD68-expressing microglia/macrophages may elicit long-term anti-inflammatory effects by secreting TGFβ. Although we did not present any results regarding neuronal recovery, numerous studies have reported a decrease in infarct volume, neuronal cell death, and behavioral deficits following intra-carotid [32], intranasal [33], intra-ventricular [34], or adenoviral [35] delivery of TGFβ in various animal models of ischemic stroke.
Moreover, we analysed the M1/M2 polarization of CD68+ cells using iNOS and CD206. Interestingly, CD206 mRNA and protein expression, in accordance with the increased expression of TGFβ, is detected at 1~3 days, peaks at 3~5 days, and gradually diminishes at 14 days after MCAO [13]. Notably, although CD206 expression decreases in the ischemic area between 5 and 14 days after ischemia, TGFβ expression increases in CD68-expressing cells. These findings suggest that most of the TGFβ is produced by CD68-expressing microglia/macrophages but not by CD206-expressing cells in the chronic stage after stroke.
iNOS is one of the isoforms of NOS, an enzyme family which includes endothelial NOS (eNOS) and neuronal NOS (nNOS) [36]. This enzyme is expressed in the macrophages/microglia, astrocytes, and various tumor cells in response to pro-inflammatory cytokines or endotoxin, and is primarily considered as a marker of M1 macrophage polarization. In our study, despite the relatively few detected iNOS+ cells, 10~34% of iNOS was co-expressed in the CD68-expressing cells in the ipsilateral hemisphere after the subacute stage, suggesting that their polarization corresponds to the M1 phenotype. According to previous studies on microglia [37, 38], these chronic M1 macrophages phagocytize the cellular debris and clear the damaged tissue in the chronic stages of stroke.
CD68 is a microglia/macrophage marker related to lysosomal glycoproteins that shuttles in vesicles between lysosomes, endosomes, and the plasma membrane [39, 40]. For this reason, various studies [41–43], in addition to our results, detected CD68 staining in the cytosol.
Our data suggest that CD68-specific cells can be considered blood-derived macrophages. Previous studies showed that blood-derived macrophages were abundantly expressed 3~7 days after MCAO, although it was difficult to distinguish them from CD68+ microglial cells [44, 45]. Subsequent studies using green fluorescent protein transgenic mice or microglia-specific markers [46] have resolved this issue [47, 48].
A study using a Rgs5gfp/+ mouse model has shown that pericytes are recruited from the blood vessels and differentiate into microglial cells [49]. In particular, activated pericytes located on the capillary wall migrate to the lesion 7 days after infarction and express several microglial markers (including CD11b, Iba1, and galectin-3) at 7~21 days after MCAO. In fact, the same study also demonstrated that the pericytes may not correlate with CD68+ cells at 1~7 days after ischemic stroke. Thus, our data collectively suggest that the proliferating CD68− (Fig. 7C) or Iba1+ microglial cell (Fig. 8) can be considered as pericyte-derived microglia/macrophages.
MRI protocols, including ADC, T1, T2, DWI, and FLAIR imaging, are highly dependent on the occlusion and reperfusion time during ischemic stroke, especially in the acute periods after ischemic cerebral infarction [50]. Therefore, suitable MRI sequences are required for use in MCAO animal models. Among these sequences, ADC and DWI exhibit high contrast in the hyperacute and acute stages of stroke but are not useful in the late stages [51]; conversely, the FLAIR sequence is a reliable method in the chronic stage [52, 53]. Consequently, in the present study we employed FLAIR imaging to measure infarct volumes and their association with immunohistochemical factors after 24 hours post-reperfusion. It should be noted that we also identified ventriculomegaly in the contralateral hemisphere on FLAIR images (data not shown). Ventriculomegaly is a common finding in patients with stroke, traumatic injury, and infectious disease, although it is difficult to determine whether ventricular enlargement is related to hydrocephalus [54]. Previous studies which analysed the contralateral hemisphere damage, such as periventricle changes from astrocytic and microglial inflammation [55] did not report any significant effects on cell death.
IRW is widely utilized for analysis of positron emission tomography data based on the threshold of contrast using radioactive isotope injections (e.g., 11C and 18F); therefore, the same approach could be applied to our MRI data obtained from a non-human primate stroke model to quantify the 3D infarct volume. Indeed, lesions were successfully detected in FLAIR images using the IRW threshold, likely because of the high contrast in four ROIs (including the whole brain, infarct lesion, brain template, and infarction template). To our knowledge, this is the first study to semi-automatically determine the infarct volume from FLAIR images by equivalent thresholds. This method may be particularly useful in the clinical setting, with applications in examinations of the chronic stage of stroke.
In the present study, moreover, we analysed the proliferation patterns of CD68+, Iba1+ macrophage/microglia, and CD13+ pericytes. Specifically, several proliferating CD68+ cells were observed in the subacute stage. Interestingly, these cells were also found at specific time points in a number of other CNS diseases. For instance, 29% of the CD68+ cells co-expressed Ki67 on day 4 after the ischemic injury [56]. Moreover, they were observed on day 7 after spinal cord injury [57], or at week 3 after the induction of hypertension [58]. Additionally, this hypertensive model showed a dramatic decrease in proliferating CD68+ cells at week 4. Since proliferating CD68+ cells have not been previously reported in the MCAO model of rhesus monkey, this expression pattern could be valuable information.
The present study provides evidence for a novel approach to treating stroke after the subacute stage (2 weeks~20 months). Specifically, because the activity of CD206+ M2-type microglia/ macrophages decreased during this time frame, activated TGFβ-secreting CD68 cells may exert anti-inflammatory effects. Furthermore, because almost all neurogenesis-promoting chemicals have poor blood-brain barrier permeability, regulation of the endogenous CD68+ cells may be an effective alternative treatment strategy. For instance, intranasal treatment with TGFβ increases the density of doublecortin-positive migrating neuroblasts and NeuN+ mature neuronal cells in the subventricular zone (SVZ) and the striatum in the MCAO mouse model [33]. This treatment also significantly reduces the infarct volume and improves functional outcomes, as measured with the modified Neurological Severity Scores test. Therefore, additional investigation of anti-inflammatory activity in the infarct area will further our understanding of the role of TGFβ-secreting CD68 cells in the brain after ischemic damage.
There were several limitations in our study. First, only two experimental animals were present in each group, for analysis, at the respective time points after stroke. Secondly, CD68 may not be representative of all macrophages. A recent report summarized various M1/M2 (or M0) type markers based on the microenvironments [59], suggesting 12 markers (CX3CR1, CD11b, CD45hi, CD45low, MERTK, MHC class IIhi, MHC class IIlow, TMEM119, P2RY12, SALL1, CD206, and CD163) that can be used to distinguish between 4 cell types (meningeal macrophages, perivascular macrophages, parenchymal endogenous microglia, and choroid plexus macrophages). Thirdly, each animal was induced different occlusion time, since it was dependent on plateaued ADC-derived volume of the focal ischemia from our previous MRI study [15]. For instance, the infarct volume of animal R326 was significantly larger than in others animals, potentially resulting in greater induction of anti-inflammatory processes. Additionally, duration of recovered infarct volume or behavioral function could be different from other reports [60, 61]. However, since our stroke model is supposed to be diffusion-perfusion matched, it is worth reducing the overestimation of the infarct core by including part of the penumbra.
The present study revealed the presence of TGFβ-secreting CD68 cells after subacute ischemic brain damage. Our findings suggest that TGFβ-secreting CD68 cells may be used as a potential therapeutic target, instead of CD206+ cells. However, further
ACKNOWLEDGEMENTS
The authors gratefully acknowledge the support of Kyu-Tae Chang at the National Primate Research Center, Korea Research Institute of Bioscience and Biotechnology, Cheongju, Republic of Korea. This research was supported by the Bio & Medical Technology Development Program of the National Research Foundation of Korea (NRF) funded by the Korean government, (NRF-2016M3A9B6902954, NRF-2016M3A9B6903268), and the Korea Research Institute of Bioscience and Biotechnology Research Initiative Program (KGM4621922, KGM5281921).
Figures
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Tables
Table 1 Experimental animals
| Group | Animal ID | Sex | *Age (y) | *Body weight (kg) | **Occlusion (duration) | ***MRI f/u | NHPSS | †Histological-analysis |
|---|---|---|---|---|---|---|---|---|
| Control (n=2) | R310 | M | 5 | 4.0 | X | X | X | O |
| R322 | F | 6 | 3.3 | X | X | X | O | |
| Acute (n=2) | R324 | F | 5 | 5.5 | 135 min | 3h | X | 3 h |
| R307 | M | 5 | 6.1 | 109 min | 24h | X | 24 h | |
| Subacute (n=2) | R326 | F | 5 | 4.9 | 82 min | 1w | X | 2 w |
| R330 | F | 4 | 5.3 | 111 min | 3w | X | 1 m | |
| Chronic (n=5) | R328 | F | 7 | 5.7 | 152 min | 4w | O | 4 m |
| R315 | M | 5 | 6.7 | 137 min | 4w | O | 20 m | |
| R305 | M | 5 | 4.6 | 135 min | 4w | O | ††Survival | |
| R306 | M | 5 | 7.0 | 128 min | 4w | O | ††Survival | |
| R313 | M | 5 | 6.2 | 162 min | 4w | O | ††Survival |
Nine rhesus monkeys were assessed at different time points following the induction of ischemic stroke (2 acute, 2 subacute, and 5 chronic) with 2 age-matched controls.
**Occlusion time was determined after evaluation of plateau (three consecutive ADC-derived lesion volumes) by MRI scans at 15-minute intervals.
***In vivo follow-up MRI (FLAIR, ADC, and DWI) was performed once a week starting 1 day post-infarct until 4 weeks after reperfusion to measure the infarct volume.
†Histological analyses were conducted by post-mortem examination after the animal’s natural death.
††Animals still surviving at the time of manuscript submission (approximately 3 years) with no corresponding MRI data.
h, hours; w, weeks; m, months; f/u, follow-up; FLAIR, fluid-attenuated inversion recovery; ADC, apparent diffusion coefficient; DWI, diffusion weighted imaging; NHPSS, non-human primate stroke scale; MRI, magnetic resonance imaging.
Table 2 3D infarct volume calculation with a threshold
| ROI | Volume (mm3) | Volume (%) | Mean | SD | Min | Max | Threshold Min/Max | |
|---|---|---|---|---|---|---|---|---|
| R326 (24h) | Whole Brain | 90615.50 | 100.00 | 57.3 | 16.0 | 38.0 | 132.5 | 38.0/251.0 |
| Infarct lesion | 10273.90 | 11.34 | 95.8 | 8.0 | 81.0 | 132.5 | 81.0/251.0 | |
| R326 (1w) | Whole Brain | 94266.70 | 100.00 | 69.6 | 20.1 | 38.0 | 148.3 | 38.0/251.0 |
| Infarct lesion | 15770.70 | 16.73 | 108.4 | 12.1 | 81.0 | 148.3 | 81.0/251.0 | |
| R330 (24h) | Whole Brain | 87765.20 | 100.00 | 65.2 | 14.1 | 38.0 | 193.0 | 38.0/251.0 |
| Infarct lesion | 3060.40 | 3.49 | 120.5 | 19.8 | 81.0 | 193.0 | 81.0/251.0 | |
| R330 (1w) | Whole Brain | 86865.00 | 100.00 | 63.8 | 12.1 | 38.0 | 152.3 | 38.0/251.0 |
| Infarct lesion | 3062.00 | 3.53 | 107.5 | 13.2 | 81.0 | 152.3 | 81.0/251.0 | |
| R330 (2w) | Whole Brain | 86345.10 | 100.00 | 66.3 | 10.7 | 3.3 | 160.1 | 38.0/251.0 |
| Infarct lesion | 1063.70 | 1.23 | 109.1 | 15.0 | 81.0 | 160.1 | 81.0/251.0 | |
| R330 (3w) | Whole Brain | 82786.50 | 100.00 | 55.4 | 7.6 | 38.0 | 128.6 | 38.0/251.0 |
| Infarct lesion | 385.40 | 0.47 | 94.3 | 9.8 | 81.0 | 128.6 | 81.0/251.0 | |
| R328 (24h) | Whole Brain | 109550.00 | 100.00 | 49.6 | 8.7 | 1.0 | 114.0 | 38.0/251.0 |
| Infarct lesion | 1260.70 | 1.15 | 90.2 | 5.9 | 81.0 | 114.0 | 81.0/251.0 | |
| R328 (1w) | Whole Brain | 107460.00 | 100.00 | 59.2 | 9.0 | 8.8 | 128.7 | 38.0/251.0 |
| Infarct lesion | 1745.40 | 1.62 | 94.7 | 7.9 | 81.0 | 128.7 | 81.0/251.0 | |
| R328 (2w) | Whole Brain | 107650.00 | 100.00 | 61.2 | 8.4 | 1.6 | 134.4 | 38.0/251.0 |
| Infarct lesion | 464.80 | 0.43 | 98.1 | 10.9 | 81.0 | 134.4 | 81.0/251.0 | |
| R328 (3w) | Whole Brain | 107520.00 | 100.00 | 63.5 | 8.9 | 3.0 | 139.3 | 38.0/251.0 |
| Infarct lesion | 400.50 | 0.37 | 96.0 | 10.6 | 81.0 | 139.3 | 81.0/251.0 | |
| R328 (4w) | Whole Brain | 105290.00 | 100.00 | 58.3 | 8.3 | 32.0 | 122.0 | 38.0/251.1 |
| Infarct lesion | 241.50 | 0.23 | 90.6 | 7.3 | 81.0 | 122.0 | 81.0/251.1 | |
| R315 (24h) | Whole Brain | 105500.00 | 100.00 | 61.6 | 14.3 | 0.0 | 151.0 | 38.0/251.0 |
| Infarct lesion | 5755.40 | 5.46 | 108.0 | 10.5 | 81.0 | 151.0 | 81.0/251.0 | |
| R315 (1w) | Whole Brain | 104140.00 | 100.00 | 55.7 | 10.6 | 1.1 | 112.0 | 38.0/251.0 |
| Infarct lesion | 4280.80 | 4.11 | 90.2 | 5.6 | 81.0 | 112.0 | 81.0/251.0 | |
| R315 (2w) | Whole Brain | 103990.00 | 100.00 | 56.8 | 9.8 | 10.7 | 136.1 | 38.0/251.0 |
| Infarct lesion | 1649.90 | 1.59 | 97.3 | 10.0 | 81.0 | 136.1 | 81.0/251.0 | |
| R315 (3w) | Whole Brain | 104270.00 | 100.00 | 58.4 | 9.8 | 0.9 | 124.3 | 38.0/251.0 |
| Infarct lesion | 1453.70 | 1.39 | 92.7 | 7.1 | 81.0 | 124.3 | 81.0/251.0 | |
| R315 (4w) | Whole Brain | 100810.00 | 100.00 | 54.5 | 8.4 | 7.1 | 98.9 | 38.0/251.1 |
| Infarct lesion | 109.20 | 0.11 | 83.9 | 3.3 | 80.1 | 98.9 | 81.0/251.1 |
The whole brain, infarct lesion, brain template, and infarction template were selected for ROIs. Infarct volume (%)=infarct lesion (mm3) / whole brain (mm3)×100 without template volume. Mean, SD, min, and max were also automatically calculated by IRW in the infarct lesion for the brain excluding the template.
References
- Mendis S, Davis S, Norrving B (2015) Organizational update: the world health organization global status report on noncommunicable diseases 2014; one more landmark step in the combat against stroke and vascular disease. Stroke 46 : e121-e122.


- Benjamin EJ, Blaha MJ, Chiuve SE, Cushman M, Das SR, Deo R, de Ferranti SD, Floyd J, Fornage M, Gillespie C, Isasi CR, Jiménez MC, Jordan LC, Judd SE, Lackland D, Lichtman JH, Lisabeth L, Liu S, Longenecker CT, Mackey RH, Matsushita K, Mozaffarian D, Mussolino ME, Nasir K, Neumar RW, Palaniappan L, Pandey DK, Thiagarajan RR, Reeves MJ, Ritchey M, Rodriguez CJ, Roth GA, Rosamond WD, Sasson C, Towfighi A, Tsao CW, Turner MB, Virani SS, Voeks JH, Willey JZ, Wilkins JT, Wu JH, Alger HM, Wong SS, Muntner P; American Heart Association Statistics Committee and Stroke Statistics Subcommittee (2017) Heart disease and stroke statistics-2017 update: a report from the American Heart Association. Circulation 135 : e146-e603.

- Davis SM, Donnan GA (2009) 4.5 hours: the new time window for tissue plasminogen activator in stroke. Stroke 40 : 2266-2267.
- Vogelgesang A, Becker KJ, Dressel A (2014) Immunological consequences of ischemic stroke. Acta Neurol Scand 129 : 1-12.
- Perego C, Fumagalli S, De Simoni MG (2011) Temporal pattern of expression and colocalization of microglia/macrophage phenotype markers following brain ischemic injury in mice. J Neuroinflammation 8 : 174.
- Lawson LJ, Perry VH, Gordon S (1992) Turnover of resident microglia in the normal adult mouse brain. Neuroscience 48 : 405-415.
- Lehrmann E, Kiefer R, Christensen T, Toyka KV, Zimmer J, Diemer NH, Hartung HP, Finsen B (1998) Microglia and macrophages are major sources of locally produced transforming growth factor-beta1 after transient middle cerebral artery occlusion in rats. Glia 24 : 437-448.
- Doyle KP, Cekanaviciute E, Mamer LE, Buckwalter MS (2010) TGFβ signaling in the brain increases with aging and signals to astrocytes and innate immune cells in the weeks after stroke. J Neuroinflammation 7 : 62.
- Lee Y, Lee SR, Choi SS, Yeo HG, Chang KT, Lee HJ (2014) Therapeutically targeting neuroinflammation and microglia after acute ischemic stroke. BioMed Res Int 2014 : 297241.
- Asai H, Ikezu S, Woodbury ME, Yonemoto GM, Cui L, Ikezu T (2014) Accelerated neurodegeneration and neuroinflammation in transgenic mice expressing P301L tau mutant and tau-tubulin kinase 1. Am J Pathol 184 : 808-818.
- Pisanu A, Lecca D, Mulas G, Wardas J, Simbula G, Spiga S, Carta AR (2014) Dynamic changes in pro- and antiinflammatory cytokines in microglia after PPAR-γ agonist neuroprotective treatment in the MPTPp mouse model of progressive Parkinson’s disease. Neurobiol Dis 71 : 280-291.
- Holder GE, McGary CM, Johnson EM, Zheng R, John VT, Sugimoto C, Kuroda MJ, Kim WK (2014) Expression of the mannose receptor CD206 in HIV and SIV encephalitis: a phenotypic switch of brain perivascular macrophages with virus infection. J Neuroimmune Pharmacol 9 : 716-726.
- Hu X, Li P, Guo Y, Wang H, Leak RK, Chen S, Gao Y, Chen J (2012) Microglia/macrophage polarization dynamics reveal novel mechanism of injury expansion after focal cerebral ischemia. Stroke 43 : 3063-3070.
- Patel AR, Ritzel R, McCullough LD, Liu F (2013) Microglia and ischemic stroke: a double-edged sword. Int J Physiol Pathophysiol Pharmacol 5 : 73-90.
- Yi KS, Choi CH, Lee SR, Lee HJ, Lee Y, Jeong KJ, Hwang J, Chang KT, Cha SH (2017) Sustained diffusion reversal with in-bore reperfusion in monkey stroke models: Confirmed by prospective magnetic resonance imaging. J Cereb Blood Flow Metab 37 : 2002-2012.
- England TJ, Sprigg N, Alasheev AM, Belkin AA, Kumar A, Prasad K, Bath PM (2016) Granulocyte-colony stimulating factor (G-CSF) for stroke: an individual patient data metaanalysis. Sci Rep 6 : 36567.
- Darusman HS, Call J, Sajuthi D, Schapiro SJ, Gjedde A, Kalliokoski O, Hau J (2014) Delayed response task performance as a function of age in cynomolgus monkeys (Macaca fascicularis). Primates 55 : 259-267.
- Moss MB, Moore TL, Schettler SP, Killiany R, Rosene D (2007) Successful vs. unsuccessful aging in the rhesus monkey. In: Brain aging: models, methods, and mechanisms (Riddle DR, ed), pp 21-38. CRC Press/Taylor & Francis, Boca Raton, FL.
- Lee Y, Kim YH, Park SJ, Huh JW, Kim SH, Kim SU, Kim JS, Jeong KJ, Lee KM, Hong Y, Lee SR, Chang KT (2014) Insulin/ IGF signaling-related gene expression in the brain of a sporadic Alzheimer's disease monkey model induced by intracerebroventricular injection of streptozotocin. J Alzheimers Dis 38 : 251-267.
- Yeo HG, Lee Y, Jeon CY, Jeong KJ, Jin YB, Kang P, Kim SU, Kim JS, Huh JW, Kim YH, Sim BW, Song BS, Park YH, Hong Y, Lee SR, Chang KT (2015) Characterization of cerebral damage in a monkey model of Alzheimer's disease induced by intracerebroventricular injection of streptozotocin. J Alzheimers Dis 46 : 989-1005.
- Roitberg B, Khan N, Tuccar E, Kompoliti K, Chu Y, Alperin N, Kordower JH, Emborg ME (2003) Chronic ischemic stroke model in cynomolgus monkeys: behavioral, neuroimaging and anatomical study. Neurol Res 25 : 68-78.
- Cook DJ, Teves L, Tymianski M (2012) Treatment of stroke with a PSD-95 inhibitor in the gyrencephalic primate brain. Nature 483 : 213-217.
- Huang L, Wong S, Snyder EY, Hamblin MH, Lee JP (2014) Human neural stem cells rapidly ameliorate symptomatic inflammation in early-stage ischemic-reperfusion cerebral injury. Stem Cell Res Ther 5 : 129.
- Klar AS, Michalak-Mićka K, Biedermann T, Simmen-Meuli C, Reichmann E, Meuli M (2018) Characterization of M1 and M2 polarization of macrophages in vascularized human dermo-epidermal skin substitutes in vivo. Pediatr Surg Int 34 : 129-135.
- Zhang Q, Atsuta I, Liu S, Chen C, Shi S, Shi S, Le AD (2013) IL-17-mediated M1/M2 macrophage alteration contributes to pathogenesis of bisphosphonate-related osteonecrosis of the jaws. Clin Cancer Res 19 : 3176-3188.
- Liu Y, D'Arceuil HE, Westmoreland S, He J, Duggan M, Gonzalez RG, Pryor J, de Crespigny AJ (2007) Serial diffusion tensor MRI after transient and permanent cerebral ischemia in nonhuman primates. Stroke 38 : 138-145.
- Hyakkoku K, Hamanaka J, Tsuruma K, Shimazawa M, Tanaka H, Uematsu S, Akira S, Inagaki N, Nagai H, Hara H (2010) Toll-like receptor 4 (TLR4), but not TLR3 or TLR9, knockout mice have neuroprotective effects against focal cerebral ischemia. Neuroscience 171 : 258-267.
- Boutin H, LeFeuvre RA, Horai R, Asano M, Iwakura Y, Rothwell NJ (2001) Role of IL-1alpha and IL-1beta in ischemic brain damage. J Neurosci 21 : 5528-5534.
- Bohacek I, Cordeau P, Lalancette-Hébert M, Gorup D, Weng YC, Gajovic S, Kriz J (2012) Toll-like receptor 2 deficiency leads to delayed exacerbation of ischemic injury. J Neuroinflammation 9 : 191.
- Narantuya D, Nagai A, Sheikh AM, Wakabayashi K, Shiota Y, Watanabe T, Masuda J, Kobayashi S, Kim SU, Yamaguchi S (2010) Microglia transplantation attenuates white matter injury in rat chronic ischemia model via matrix metalloproteinase- 2 inhibition. Brain Res 1316 : 145-152.
- Woo MS, Park JS, Choi IY, Kim WK, Kim HS (2008) Inhibition of MMP-3 or -9 suppresses lipopolysaccharide-induced expression of proinflammatory cytokines and iNOS in microglia. J Neurochem 106 : 770-780.
- Gross CE, Bednar MM, Howard DB, Sporn MB (1993) Transforming growth factor-beta 1 reduces infarct size after experimental cerebral ischemia in a rabbit model. Stroke 24 : 558- 562.
- Ma M, Ma Y, Yi X, Guo R, Zhu W, Fan X, Xu G, Frey WH 2nd, Liu X (2008) Intranasal delivery of transforming growth factor-beta1 in mice after stroke reduces infarct volume and increases neurogenesis in the subventricular zone. BMC Neurosci 9 : 117.
- McNeill H, Williams C, Guan J, Dragunow M, Lawlor P, Sirimanne E, Nikolics K, Gluckman P (1994) Neuronal rescue with transforming growth factor-beta 1 after hypoxicischaemic brain injury. Neuroreport 5 : 901-904.
- Pang L, Ye W, Che XM, Roessler BJ, Betz AL, Yang GY (2001) Reduction of inflammatory response in the mouse brain with adenoviral-mediated transforming growth factor-ss1 expression. Stroke 32 : 544-552.
- Garry PS, Ezra M, Rowland MJ, Westbrook J, Pattinson KT (2015) The role of the nitric oxide pathway in brain injury and its treatment--from bench to bedside. Exp Neurol 263 : 235-243.
- Benakis C, Garcia-Bonilla L, Iadecola C, Anrather J (2015) The role of microglia and myeloid immune cells in acute cerebral ischemia. Front Cell Neurosci 8 : 461.
- Fumagalli S, Perego C, Pischiutta F, Zanier ER, De Simoni MG (2015) The ischemic environment drives microglia and macrophage function. Front Neurol 6 : 81.
- Holness CL, Simmons DL (1993) Molecular cloning of CD68, a human macrophage marker related to lysosomal glycoproteins. Blood 81 : 1607-1613.
- Fambrough DM, Takeyasu K, Lippincott-Schwarz J, Siegel NR (1988) Structure of LEP100, a glycoprotein that shuttles between lysosomes and the plasma membrane, deduced from the nucleotide sequence of the encoding cDNA. J Cell Biol 106 : 61-67.
- Brettschneider J, Toledo JB, Van Deerlin VM, Elman L, Mc-Cluskey L, Lee VM, Trojanowski JQ (2012) Microglial activation correlates with disease progression and upper motor neuron clinical symptoms in amyotrophic lateral sclerosis. PLoS One 7 : e39216.
- Matsui H, Ohgomori T, Natori T, Miyamoto K, Kusunoki S, Sakamoto K, Ishiguro N, Imagama S, Kadomatsu K (2013) Keratan sulfate expression in microglia is diminished in the spinal cord in experimental autoimmune neuritis. Cell Death Dis 4 : e946.
- Hendrickx DAE, van Eden CG, Schuurman KG, Hamann J, Huitinga I (2017) Staining of HLA-DR, Iba1 and CD68 in human microglia reveals partially overlapping expression depending on cellular morphology and pathology. J Neuroimmunol 309 : 12-22.
- Schilling M, Besselmann M, Müller M, Strecker JK, Ringelstein EB, Kiefer R (2005) Predominant phagocytic activity of resident microglia over hematogenous macrophages following transient focal cerebral ischemia: an investigation using green fluorescent protein transgenic bone marrow chimeric mice. Exp Neurol 196 : 290-297.
- Schilling M, Strecker JK, Schäbitz WR, Ringelstein EB, Kiefer R (2009) Effects of monocyte chemoattractant protein 1 on blood-borne cell recruitment after transient focal cerebral ischemia in mice. Neuroscience 161 : 806-812.
- Satoh J, Kino Y, Asahina N, Takitani M, Miyoshi J, Ishida T, Saito Y (2016) TMEM119 marks a subset of microglia in the human brain. Neuropathology 36 : 39-49.
- Schilling M, Besselmann M, Leonhard C, Mueller M, Ringelstein EB, Kiefer R (2003) Microglial activation precedes and predominates over macrophage infiltration in transient focal cerebral ischemia: a study in green fluorescent protein transgenic bone marrow chimeric mice. Exp Neurol 183 : 25-33.
- Tanaka R, Komine-Kobayashi M, Mochizuki H, Yamada M, Furuya T, Migita M, Shimada T, Mizuno Y, Urabe T (2003) Migration of enhanced green fluorescent protein expressing bone marrow-derived microglia/macrophage into the mouse brain following permanent focal ischemia. Neuroscience 117 : 531-539.
- Özen I, Deierborg T, Miharada K, Padel T, Englund E, Genové G, Paul G (2014) Brain pericytes acquire a microglial phenotype after stroke. Acta Neuropathol 128 : 381-396.
- European Stroke Organisation (ESO) Executive Committee; ESO Writing Committee (2008) Guidelines for management of ischaemic stroke and transient ischaemic attack 2008. Cerebrovasc Dis 25 : 457-507.
- Moseley ME, Kucharczyk J, Mintorovitch J, Cohen Y, Kurhanewicz J, Derugin N, Asgari H, Norman D (1990) Diffusion-weighted MR imaging of acute stroke: correlation with T2-weighted and magnetic susceptibility-enhanced MR imaging in cats. AJNR Am J Neuroradiol 11 : 423-429.
- Gauvrit JY, Leclerc X, Girot M, Cordonnier C, Sotoares G, Henon H, Pertuzon B, Michelin E, Devos D, Pruvo JP, Leys D (2006) Fluid-attenuated inversion recovery (FLAIR) sequences for the assessment of acute stroke: inter observer and inter technique reproducibility. J Neurol 253 : 631-635.
- Ricci PE, Burdette JH, Elster AD, Reboussin DM (1999) A comparison of fast spin-echo, fluid-attenuated inversionrecovery, and diffusion-weighted MR imaging in the first 10 days after cerebral infarction. AJNR Am J Neuroradiol 20 : 1535-1542.
- Osuka S, Matsushita A, Yamamoto T, Saotome K, Isobe T, Nagatomo Y, Masumoto T, Komatsu Y, Ishikawa E, Matsumura A (2010) Evaluation of ventriculomegaly using diffusion tensor imaging: correlations with chronic hydrocephalus and atrophy. J Neurosurg 112 : 832-839.
- Garbuzova-Davis S, Haller E, Williams SN, Haim ED, Tajiri N, Hernandez-Ontiveros DG, Frisina-Deyo A, Boffeli SM, Sanberg PR, Borlongan CV (2014) Compromised blood-brain barrier competence in remote brain areas in ischemic stroke rats at the chronic stage. J Comp Neurol 522 : 3120-3137.
- Boddaert J, Bielen K,'s Jongers B, Manocha E, Yperzeele L, Cras P, Pirici D, Kumar-Singh S (2018) CD8 signaling in microglia/ macrophage M1 polarization in a rat model of cerebral ischemia. PLoS One 13 : e0186937.
- You T, Bi Y, Li J, Zhang M, Chen X, Zhang K, Li J (2017) IL-17 induces reactive astrocytes and up-regulation of vascular endothelial growth factor (VEGF) through JAK/STAT signaling. Sci Rep 7 : 41779.
- Koizumi T, Taguchi K, Mizuta I, Toba H, Ohigashi M, Onishi O, Ikoma K, Miyata S, Nakata T, Tanaka M, Foulquier S, Steinbusch HW, Mizuno T (2019) Transiently proliferating perivascular microglia harbor M1 type and precede cerebrovascular changes in a chronic hypertension model. J Neuroinflammation 16 : 79.
- Li Q, Barres BA (2018) Microglia and macrophages in brain homeostasis and disease. Nat Rev Immunol 18 : 225-242.
- Rewell SS, Churilov L, Sidon TK, Aleksoska E, Cox SF, Macleod MR, Howells DW (2017) Evolution of ischemic damage and behavioural deficit over 6 months after MCAo in the rat: Selecting the optimal outcomes and statistical power for multi-centre preclinical trials. PLoS One 12 : e0171688.