Articles

Article Tools
 View Full Text View Full Text |
 Abstract Abstract |
 Article as PDF Article as PDF |
 Print this Article Print this Article |
 Pubmed Pubmed |
 PMC PMC |
 PubReader PubReader |
 Export to Citation Export to Citation |
 Email Alerts Email Alerts |
 Open Access Open Access |
Share this article on :



Stats or Metrics
Article
Review Article
Exp Neurobiol 2019; 28(6): 643-657
Published online November 27, 2019
https://doi.org/10.5607/en.2019.28.6.643
© The Korean Society for Brain and Neural Sciences
Autophagy, Cellular Aging and Age-related Human Diseases
So Yeong Cheon1,2, Hyunjeong Kim1,2, David C. Rubinsztein1,3* and Jong Eun Lee2,4*
1Department of Medical Genetics, Cambridge Institute for Medical Research (CIMR), University of Cambridge, Cambridge CB2 0XY, United Kingdom, 2Department of Anatomy, Yonsei University College of Medicine, Seoul 03722, Korea, 3UK Dementia Research Institute, University of Cambridge, Cambridge CB2 0AH, United Kingdom, 4BK21 PLUS Project for Medical Science, and Brain Research Institute, Yonsei University College of Medicine, Seoul 03722, Korea
Correspondence to: *To whom correspondence should be addressed.
David C. Rubinsztein, TEL: 44-1223-762608, FAX: 44-1223-331206
e-mail: dcr1000@cam.ac.uk
Jong Eun Lee, TEL: 82-2-2228-1646, FAX: 82-2-365-0700
e-mail: jelee@yuhs.ac
This is an Open Access article distributed under the terms of the Creative Commons Attribution Non-Commercial License
(http://creativecommons.org/licenses/by-nc/4.0) which permits unrestricted non-commercial use, distribution, and
reproduction in any medium, provided the original work is properly cited.
Abstract
Macroautophagy/autophagy is a conserved degradation system that engulfs intracytoplasmic contents, including aggregated proteins and organelles, which is crucial for cellular homeostasis. During aging, cellular factors suggested as the cause of aging have been reported to be associated with progressively compromised autophagy. Dysfunctional autophagy may contribute to age-related diseases, such as neurodegenerative disease, cancer, and metabolic syndrome, in the elderly. Therefore, restoration of impaired autophagy to normal may help to prevent age-related disease and extend lifespan and longevity. Therefore, this review aims to provide an overview of the mechanisms of autophagy underlying cellular aging and the consequent disease. Understanding the mechanisms of autophagy may provide potential information to aid therapeutic interventions in age-related diseases.
Graphical Abstract
Keywords: Autophagy, Aging, DNA damage, Oxidative stress, Telomere shortening, SASP
INTRODUCTION
Aging causes biological and functional changes not only in organisms but also at the cellular level [1–3]. Failure of repair systems progresses to cellular damage, which drives the aging process. Oxidative stress, DNA damage, telomere shortening and inflammatory senescence-associated secretory phenotype (SASP) may be prominent causative agents of aging [4–7]. These age-related factors can adversely affect normal cellular homeostasis, which may contribute to the development or progression of age-related diseases, including neurodegenerative conditions (Alzheimer’s disease, Huntington’s disease, Parkinson’s disease, and amyotrophic lateral sclerosis) [8–11].
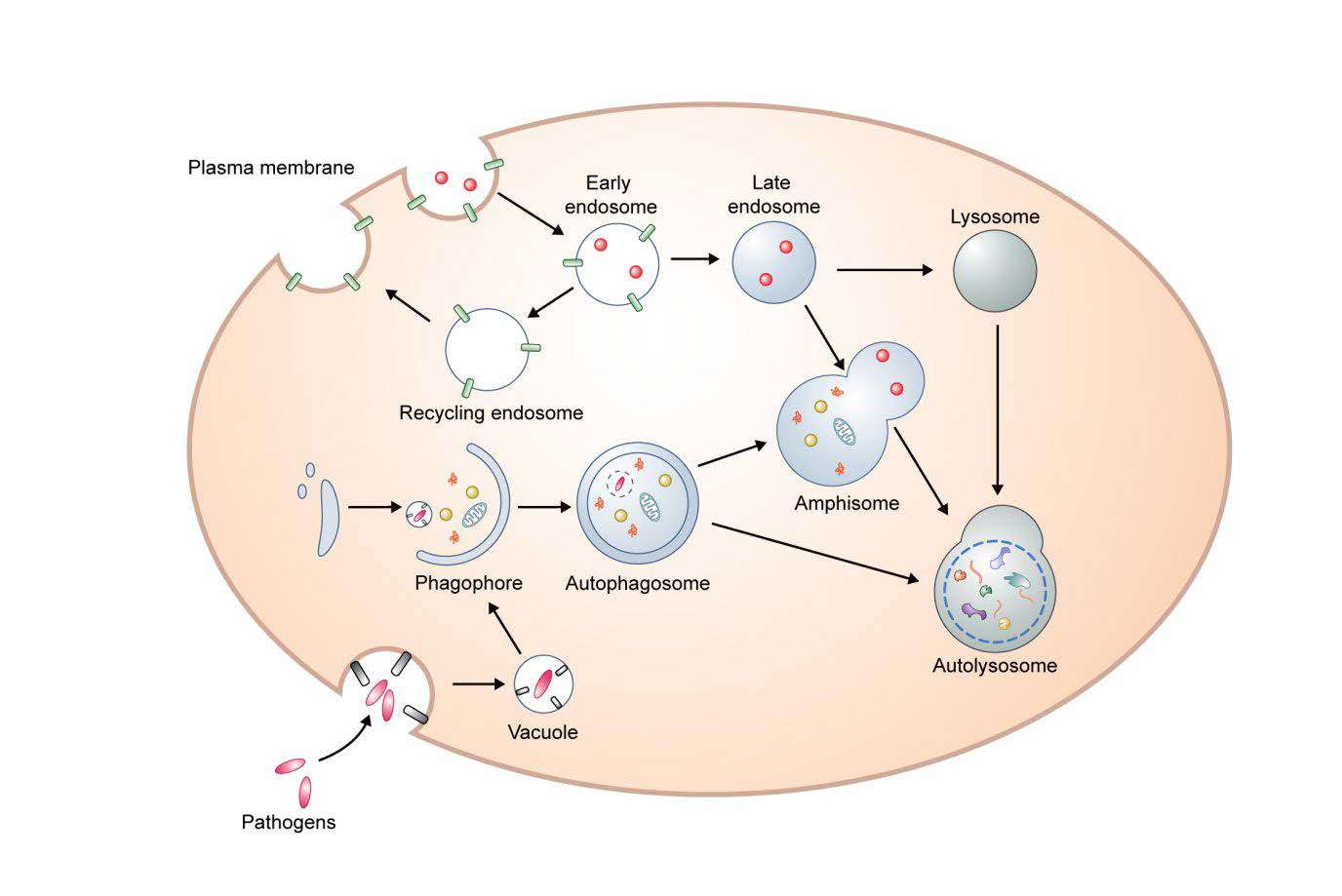
Macroautophagy (herein autophagy) is a conserved intracellular degradation mechanism that delivers cytoplasmic contents, such as aggregated proteins and organelles, to the lysosome for clearance. Autophagosomes derive from phagophores, cup-shaped and double-membrane structures. After the edges of the phagophore extend and fuse, the resulting autophagosomes have captured cytoplasmic proteins and organelles. Autophagosomes ultimately fuse with lysosomes to become autolysosomes, enabling the degradation of the autophagic contents. (Fig. 1). During starvation, this process is in general involved in cell survival and cellular energy formation by enabling nutrient recycling [12, 13]. Amino acids negatively regulate autophagy via mammalian target of rapamycin (mTOR) signaling, a negative regular of autophagy [14]. Under non-starvation and starvation conditions, autophagy can degrade aggregate-prone proteins (aggrephagy), intracellular pathogens (xenophagy), damaged mitochondria (mitophagy), excess peroxisomes (perophagy), and impaired endoplamic reticulum (ER-phagy) [12, 15].
During normal aging, the expression of some autophagy-associated genes, including autophagy-related (ATG) protein 5 (ATG5), ATG7, and beclin1, may decline in the human brain [16]. Autophagy gene polymorphisms have been implicated in age-related neurodegeneration, suggesting that impairment or defects in the autophagy machinery may contribute to age-related pathologies [13, 17, 18]. Intracellular accumulation of protein aggregates due to compromised autophagy is common in pathological conditions [12]. Restoring the deterioration of autophagy may be a rational strategy for improving aging-related diseases [1]. In this review, we aim to summarize how autophagy is linked to aging factors in cellular pathology and age-related disease.
OVERVIEW OF AUTOPHAGY PROCESS
One of the earliest steps regulating autophagosome formation is the activation of the Unc-51-like kinase (ULK) complex that comprises ULK1, ULK2, ATG13, focal adhesion kinase (FAK) family-interacting protein of 200kDa (FIP200), and ATG101 [13]. The ULK complex is activated by 5′-adenosine monophosphate (AMP)-activated protein kinase (AMPK) or inhibited by mTOR complex 1, which regulate ULK phosphorylation [12, 19]. Subsequently, this complex recruits the class III phosphatidylinositol 3-kinase (PI3K) complex I that contains VPS34, VPS15, beclin1, and ATG14L. This complex enables phosphatidylinositol 3-phosphate (PI3P) generation on the phagophore [1, 12]. PI3P induces the recruitment of a PI3K-binding proteins, tryptophan-aspartic acid (WD) repeat domain phosphoinositide-interacting (WIPI 1 and 2) [13]. WIPI2 can directly bind to ATG16L1, thereby allowing it to be recruited by the ATG12-ATG5-ATG16L1 complex [20]. ATG12 is conjugated to ATG5 via ATG7 (E1-like enzyme) and ATG10 (E2-like enzyme), and ATG12-ATG5 conjugates can bind to ATG16L1 non-covalently (ATG12-ATG5-ATG16L1) [12]. The microtubule-associated protein 1 light chain3 (LC3) family members, like LC3 proteins, are cleaved in their C-terminal tails by ATG4. Cleaved LC3 (LC3-I) can be conjugated in an ubiquitin-like manner to the lipid phosphatidylethanolamine (PE) on phagophore membranes via ATG7 (E1-like enzyme), ATG3 (E2-like enzyme) [12, 21], and the E3 like-ATG12-ATG5-ATG16L1. The lipidated LC3 is known as LC3-II. LC3 family members conjugated to phagophores are thought to contribute to membrane elongation, closure, and recruitment of autophagy receptors, such as p62/sequestosome (SQSTM1) and optineurin, which assist in the recruitment of various selective autophagy substrates to phagophores [1, 12].
Autophagosomal membranes may derive from a number of sources including ER, Golgi, mitochondria, endosomes and the plasma membrane [22]. Mammalian ATG9 and ATG16L1 traffic from the plasma membrane to sites of autophagosome formation [23]. Homotypic fusion of early ATG16L1 vesicles mediated by the soluble N-ethylmaleimide sensitive factor attachment protein receptors (SNAREs) protein vesicle-associated membrane protein (VAMP)7, contributes to phagophore genesis [24]. Interestingly, mATG9-contating vesicles can meet ATG16L1 vesicles in recycling endosomes and heterotypic fusion between mATG9 and ATG16L1 is mediated the by SNARE protein VAMP3 [25], and these processes are important for autophagosome biogenesis. In addition, it has been suggested that the SNARE protein syntaxin 17 (STX17) on the ER binds ATG14L and relocates ATG14L to ER-mitochondria contact sites, implicating ER-mitochondria contact sites in autophagosome formation [26]. Recent data suggest that many of the key events in autophagosome formation, including LC3 conjugation to PE, occur on RAB11A-positive recycling endosomes [27].
Completed autophagosomes migrate along microtubules toward the perinuclear localization where lysosomes are concentrated [13]. The minus-end-directed dynein and plus-end-directed kinesin are key motor proteins that transport autophagosomes [28–30]. Before forming autolysosomes, autophagosomes can fuse with endosomes resulting in amphisomes [31].
Membrane fusion requires a series of tethering and docking processes [32]. Homotypic fusion and vacuole protein sorting (HOPS) complex, ectopic P granules protein 5 (EPG5), and pleckstrin homology domain containing protein family member 1 (PLEKHM1) have been suggested as tethering factors for autophagosome-lysosome fusion [33]. After tethering, autophagosome-lysosome fusion is mediated by SNAREs [34]. Through these processes, autophagosomes eventually undergo fusion with acidic lysosomes for degradation (autolysosome) [12, 13].
AGE-RELATED FACTORS AND AUTOPHAGY
Oxidative stress and autophagy
Oxidative stress caused by reactive oxygen species (ROS) including several types of free radicals (superoxide (O2•−), hydrogen peroxide (H2O2), and hydroxyl radicals (OH•)) is thought to affect cellular aging [4, 35]. The main antioxidant enzymes, such as superoxide dismutase (SOD), catalase (CAT), peroxidase (GPX) and glutathione (GSH), protect against excessive ROS, but their function is disrupted by cellular aging [4, 35]. Intracellular ROS is mainly generated by the electron transport chain (ETC) in mitochondria. The ETC transfers electrons via an electrochemical gradient (Δp), consisting of a membrane potential (Δψ) and a pH gradient (ΔpH), that synthesizes adenosine triphosphate (ATP) by the process of oxidative phosphorylation (OXPHOS) across mitochondrial inner membrane [36, 37]. During this process, electrons from nicotinamide adenine dinucleotide (NADH) at the complex I (NADH dehydrogenase) site and FADH2 at the complex II (succinate dehydrogenase) site are transferred by coenzyme Q, which transports electrons to complex III (cytochrome
Under normal conditions, ROS can regulate autophagy. However, excessive ROS can impair organelles and lead to protein modification and aggregation. Conversely, autophagy can decrease oxidative damage. O2•− is induced by starvation, or a lack of pyruvate, L-glutamine, or glucose. Both O2•− and H2O2 are induced by starvation. Mitochondria-generated O2•− and H2O2 are major autophagy regulators [42, 45–47]. Under starvation conditions, ROS-induced activation of AMPK induces autophagy [48]. In mETC deficient cells, O2•− production is blocked under starvation conditions, thereby reducing the activation of AMPK and increasing activation of the mTOR pathway, which results in a reduction of starvation-induced autophagy [48]. Downstream of AMPK, peroxisome proliferator-activated receptor gamma coactivator 1-alpha (PGC-1α), is required for modulation of antioxidant genes in response to oxidative stress. The AMPK-PGC-1α signaling pathway controls mitochondrial ROS. Cells with reduced AMPK activity increase mitochondrial ROS and experience premature aging [49]. Parkinsonism-causing genes like Parkin, phosphatase and tensin homolog-induced putative kinase (PINK), and DJ-1 impact mitochondrial health via autophagy [50–52]. ROS promotes Parkin/PINK-dependent mitophagy [50]. Also, Parkin induces beclin1-mediated autophagic degradation of molecular debris and dysfunctional mitochondria, which prevents oxidative stress [51]. Loss of PINK1 compromises the function of mitochondrial complexes I and II, and increases sensitivity to oxidative stress with aging [53]. Similar to Parkin and PINK1, DJ-1, a redox-dependent molecular chaperone, is also associated with mitochondria [54]. The loss of DJ-1 increases fragmentation of mitochondria, causes accumulation of autophagy-associated factors, such as LC3, and lowers mitochondrial membrane potential [52]. On the other hand, overexpression of DJ-1 suppresses formation of protein aggregates. Because DJ-1 is genetically associated with Parkin and PINK1, it can rescue the effects of PINK1 mutation in
DNA damage and autophagy
Extrinsic agents, such as ultraviolet (UV) light and toxins, or intrinsic stimuli such as ROS can cause damage to DNA. Damaged DNA is associated with cellular dysfunction. A declining ability to repair DNA and consequent accumulation of DNA damage may contribute to cellular senescence. Also, mutations in nuclear and mitochondrial genes caused by impaired DNA repair have been associated with aging. DNA damage includes mismatch, strand breaks (single or double), and base modification, which contribute to DNA lesions. Numerous DNA repair systems are activated in response to damaged DNA, including homologous recombination repair (HR), non-homologous end joining (NHEJ), mismatch repair (MMR) base excision repair (BER), and nucleotide excision repair (NER) [56, 57]. HR accurately repairs harmful double strand breaks (DSBs) in DNA using a homologous DNA template. NHEJ also repairs DSBs and ligates broken ends directly (non-homologous). MMR detects and repairs erroneous base incorporation and insertions/deletions. BER recognizes and eliminates non-bulky lesions in DNA caused by apurinic/apyrimidinic sites (AP sites), oxidation, deamination, and alkylation. In the case of NER, it removes bulky or helix-distorting DNA lesions, which come from UV light, chemicals, and radiation [56, 58–60]. Many previous studies have implicated defective DNA repair system in aging [5, 61]. In an
Interestingly, it has been reported that DNA repair is closely connected to autophagy [63]. Beclin1 hemizygosity compromises autophagy and activates DNA damage responses in mammary epithelial cells [64]. Likewise, defective autophagy leads to impaired DNA repair. In particular, accumulation of p62 has been observed after loss of autophagy, which interrupts DNA damage responses (DDR) [65]. Inhibition of p62 accumulation mitigates defective autophagy-induced genome damage [66]. The p62 is involved in regulation of NHEJ and HR. In a p62-dependent manner, recruitment of DNA repair proteins, such as BRCA1, RAP80 and Rad51, to double strand breaks is compromised under autophagic deficiency (Atg3 knockout) [65]. Interestingly, the balance between HR and NHEJ is regulated by p62 [67]. In addition, suppression of NHEJ causes DNA damage and cell death in an autophagy-deficient environment (Atg7 null cells). Autophagy-deficient cells have defective activation of checkpoint kinase-1 (Chk1), which contributes to DNA repair by HR. Its failure in response to DNA damage leads to reduced recruitment of Rad51 to the damaged sites, which is important to correct HR [68]. Furthermore, autophagy mediates NER by increasing recognition of damaged DNA. In Atg5-deficient embryonic fibroblast cells, autophagy deficiency results in the failure to sense damaged DNA by XP group C (XPC) and DNA damaging-binding protein 2 (DDB2), which are important in recognition of global genome NER-specific damage after UV-induced stress [69]. Also, impaired autophagy results in DNA damage, increased mutation rates and chromosomal instability in breast epithelial cells with allelic loss of beclin1or in Atg5 null mouse embryonic fibroblasts (MEFs) [70].
Mitochondrial DNA (mtDNA) is more vulnerable to ROS than nuclear DNA [71, 72]. Mutation of mtDNA is likely due to replication errors by mtDNA polymerase and point mutations/deletions accumulate with aging. mtDNA deletions are more frequent in the aging brain [73]. Previous studies support that the BER is mainly involved in repair of oxidative mtDNA modification and mitigate mitochondrial impairment [74]. MMR-dependent autophagy requires Bcl-2 Interacting Protein 3 (BNIP3) in an mTOR-dependent manner and BNIP3 is important in mitophagy in response to hypoxia [75, 76].
Telomere shortening and autophagy
Telomeres are composed of six base pair repeated DNA sequences (TTAGGG) at the end of linear chromosomes. These protect genetic information during DNA replication and protect chromosomal ends from damage or being recognized as DNA breaks, thereby maintaining genomic stability [6, 77]. Telomeres bind to the shelterin complex, which includes telomere-binding proteins, such as protection of telomeres protein-1 (POT1), telomeric repeat binding factor-1 (TRF1), TRF2, TRF1-interacting protein-2 (TIN2), repressor/activator protein1 (RAP1), and TIN2- and POT-1interacting protein (TPP1). These components generate t-loops and regulate telomere integrity and synthesis of telomeric DNA by telomerase. Furthermore, these proteins interact with DNA repair factors and protect telomeres from damage [78]. To maintain telomere length, a telomerase consisting of telomerase RNA competent (TERC) and telomerase reverse transcriptase (TERT), helps to add telomeric DNA at the 3′ ends of telomeres. However, telomeres progressively shorten with cell division/DNA replication cycle, which is caused by the lack of telomerase activity. Normally, aging leads to functional decline of telomerase in most of somatic cells. However, in immortalized cells, such as germ cells and cancer cells, telomerase can sustain its activity. It is difficult to restore telomeres with the DNA repair machinery and this eventually results in cellular senescence and aging [6, 77, 79].
Aged telomerase-deficient mice show impaired telomere function, increased genetic instability, and a higher incidence of cancer [80]. Similarly, Terc deficiency results in short telomeres in older mice. Conversely, Terc haploinsufficiency rescues aberrant telomeres and prevents premature aging [81]. In
Previous studies have shown that telomere biology is associated with autophagy [87–89]. In cells undergoing crisis, which is associated with deprotected telomeres, there are increased cytoplasmic vacuoles and autophagy-related proteins (ATG5-ATG12, LC3-II) [88]. Telomeric 3′ overhang-specific DNA oligonucleotides, which mimic telomere loop disruption, can result in induction of autophagosomes and inhibit mTOR signaling in malignant glioma cells [87]. In addition, in multiple cell lines, such as HEK 293T, HepG2, and U-2 OS, TERT binds to and suppresses mTORC1 kinase, thereby inducing autophagy. TERT knockdown increases components of mTORC1, resulting autophagy impairment under basal and amino acid starvation conditions [89]. Autophagy contributes to inhibition of p53, which can be affected by telomere shortening. As a feedback mechanism, p53 can activate autophagy [85, 90].
Inflammation and autophagy
Normally, senescent cells are eliminated by the immune system. However, this system deteriorates with aging and the accumulation of senescent cells can impair tissue homeostasis [7]. Senescence-associated secretory phenotype (SASP) is a process where senescent cells release pro-inflammatory factors (interleukin (IL)-6, IL-8, and IL-1α), proteases (matrix metallopeptidase (MMP)-1, -3, and -13), chemokines (C-C motif chemokine ligand 2 (CCL2), and macrophage inflammatory proteins (MIPs)), and growth factors (transforming growth factor-β (TGF-β)) [7, 91, 92]. SASP factors are related to migration and recruitment of immune cells and influence tissue repair in the early phase; persistent SASP from senescent cells can lead to chronic inflammation and tissue dysfunction [93]. This is accomplished by the nuclear factor kappa light chain enhancer of activated B cells (NF-κB) or p38 mitogen-activated protein kinase (MAPK) signaling pathways, upstream of SASP. Cyclic GMP-AMP synthase (cGAS)/stimulator of interferon genes (STING) pathways have been suggested as a mediator of SASP [91, 92, 94]. Through paracrine (non-cell-autonomous) or autocrine (cell-autonomous) mechanisms, SASP components from senescent cells, can reinforce cellular senescence and are involved in the progression of aging [7, 91, 95].
SASP appears to increase with aging [96–100]. For example, with age, plasma tumor necrosis factor-α (TNF-α) and IL-8 are overexpressed [96–98]. In old human fibroblasts, cellular senescence is involved in the activation of TGF-β [99]. Also, increased MMP-1 expression is closely associated with endothelial cell senescence [100]. In human BJ fibroblasts, senescent cells show increased levels of monocyte chemotactic protein-1 (MCP-1), IL-15, IL-1β, Toll-like receptor (TLR)4, and intercellular adhesion molecule-1 (ICAM-1) [101]. In human IMR-90 fibroblasts, IL-1α signaling regulates SASP components (IL-6 and IL-8) and potentiates cellular senescence [95]. Interestingly, in the same human IMR-90 fibroblasts, CXCR2 expression is increased during replicative senescence, compared to early and middle passage cells, and deletion of CXCR2 extends lifespan of the cells [102]. In particular, ablation of cGAS reduces CCL7, VCAM-1, CCL2 and CSF1 mRNA levels. Similarly, STING knockout also diminishes CCL7 and CCL2 in MEFs. Production of some SASP components, such as IL-6 and TNF-α, is dependent on cGAS. Thus, the cGAS-STING pathway positively promotes senescence and SASP [103].
Numerous studies suggest the autophagy machinery interacts with and controls the immune system and inflammation. Also, impairment of autophagy is associated with various inflammatory diseases [104, 105]. Rapamycin, an mTOR inhibitor as well as an autophagy inducer, can reduce SASP secretion products like IL-6 and other cytokines via mTORC1, and decrease the NF-κB-IL1A positive feedback loop [106]. Knockdown of ATG5 or ATG7 can delay production of SASP components such as IL-6 and IL-8 [107]. Under autophagy-deficient conditions, p62 accumulates and induces NF-κB activity [104]. Suppression of autophagy in basal conditions induces IL-1β in human peripheral blood mononuclear cells (PBMCs) in response to TLR2 or 4 ligands. Autophagy induction under starvation conditions can lead to a reduction of IL-1β. In this process, cytokine production is dependent on p38 MAPK [108]. Similarly, macrophages from ATG16L1-deficient mice have increased production of IL-18 and IL-1β. Also, ATG7-deficient macrophages and ATG4BC74A mutated RAW264.7 macrophage cell lines manifest increased production of IL-1β following lipopolysaccharide (LPS) stimulation [109]. GATA4, as a positive regulator of SASP and senescence, triggers NF-κB activation. However, ablation of GATA4 suppresses its activity and SASP genes. The activity of the transcription factor GATA4 upregulates the genes encoding C-X-C motif ligand (CXCL) 1, IL-6, IL-8, chemokine (C-C motif) ligand (CCL) 2, and granulocyte-macrophage colony-stimulating factor (GM-CSF). Deletions of ATG5 and ATG7 or the autophagy adaptor p62 increase GATA4 levels in IMR-90 fibroblasts. Interestingly, p62 physically interacts with GATA4, an interaction which is decreased with aging. In aging human brain, increased GATA4 levels are seen in prefrontal cortex [110]. Taken together, these reports suggest that autophagy plays an important role in SASP, which is associated with aging.
AGE-RELATED DISEASES AND AUTOPHAGY
Alzheimer’s disease
In Alzheimer’s disease (AD) animal models, autophagy has protective effects against depositions of misfolded amyloid beta (Aβ) and neurofibrillary tangles from tau hyperphosphorylation, which can accelerate impairment of cell function and homeostasis [111]. Autophagy triggers Aβ and tau clearance as autophagy substrates, while autophagic failure can increase intracellular Aβ [111]. In AD, at least in later stages of disease, there is an accumulation of autophagosomes, suggesting impairment of autophagosome clearance [112]. Familial AD can be caused by mutations in Presenilin 1 (PS1), which is a γ-secretase component and is important in lysosomal acidification in autophagy [112, 113]. Increased levels of hyperactivated mTOR are observed in postmortem brains of AD [113–115]. Defects in autophagy can exacerbate cognitive impairment in an AD model [116]. However, enhancement of autophagy reduces AD-related signs in animal models. Treatment with rapamycin results in a reduction of Aβ deposition, and extended longevity in AD models [1, 111, 117]. In Tg2567 mice, rapamycin reduces Aβ burden and restores synaptic and cognitive function [118]. Trehalose, an mTOR-independent autophagy enhancer, decreases tau aggregates in a neuronal cell model of tauopathy [119], and suppresses Aβ aggregates [120].
Age-associated SASP and oxidative stress are also closely associated with AD, suggesting that these factors may contribute to its pathology [8, 121]. AD models display reactive microglia near the Aβ plaque and reactive microglia release proinflammatory cytokines, including TNF-α, IL-1β, and IL-6 [122, 123]. During aging, chronic inflammation can lead to increased amyloid precursor protein (APP), tau phosphorylation, and memory impairment [124]. AD patients have increased levels of TNF-α, ILs (IL-1β, IL-6, IL-12, and IL-18) and TGF-β in peripheral blood, and TGF-β in the cerebrospinal fluid (CSF), suggesting that the inflammatory immune system is closely related to the pathology and symptoms of AD [125]. Interestingly, myeloid ATG5 deficiency accelerates these cytokine levels and reactive microgliosis in the 5XFAD AD mouse model [123]. Microglia from Beclin1−/− mice have increased levels of IL-1β and IL-18 [126]. On the contrary, enhancement of autophagy with rapamycin treatment reduces the SASP [106]. Also, rapamycin inhibits Aβ25-35-induced inflammatory mediators such as TNF-α and IL-1β [127].
Oxidative stress is closely related to Aβ [128]. Aβ can cause oxidative stress [128]. Inversely, oxidative stress can accelerate Aβ deposition [129, 130]. Oxidative stress induced by H2O2 increases intracellular Aβ in the human SH-SY5Y neuroblastoma cell line [130]. Aβ may enter into the mitochondria, generate free radicals and cause mitochondrial dysfunction. In particular, Aβ inhibits mitochondrial respiration. Additionally, mutation of APP or soluble Aβ results in mitochondrial oxidative damage, which affects AD development and progression [131]. Interestingly, Parkin decreases intracellular Aβ and triggers beclin1-dependent autophagy. Parkin overexpression also blocks Aβ-induced mitochondrial dysfunction and oxidative stress. Moreover, reduced levels of beclin1 are linked to aging and neurodegenerative diseases [51]. APP transgenic mice lacking beclin1 have increased Aβ deposition, and abnormal neuronal structure [132]. On the contrary, rapamycin-induced autophagy reduces hippocampal neuronal damage in Aβ-injected rats. At the same time, it normalizes ROS and antioxidant (SOD, GSH, catalase) levels [117]. In SH-SY5Y neuroblastoma, rapamycin alleviates Aβ-induced redox imbalances, lowers ROS and increases antioxidant enzymes [133].
Huntington’s disease
Huntington’s disease (HD) is caused by abnormally long CAG trinucleotide expansions in the Huntingtin (htt) gene, encoding abnormal long polyglutamine (polyQ) tracts [134]. Indeed, HD patients show somatic instability of CAG repeats [135]. Mutated htt impairs the autophagy machinery, which results in the accumulation of mutant htt and cell death [136, 137]. On the other hand, mutant htt is an autophagy substrate and autophagy inducers, like rapamycin, induce clearance of mutant htt and reduce cell death [138, 139].
Several lines of evidence support that oxidative stress and DNA repair systems are related to mutant htt or expanded CAG repeats [9, 140]. Oxidative damage can lead to CAG expansion at the human
Parkinson’s disease
Parkinson’s disease (PD) can be caused by genetic mutations in several genes, such as autosomal dominant genes (leucine-rich repeat kinase2 [LRRK2], alpha-synuclein [SNCA], glucocerebrosidase [GBA]) and autosomal recessive genes (DJ-1, PRKN, PINK1, and ATP13A2). These genes are closely related to autophagy, including mitophagy, and autophagy failure may be an important driver of pathology in many forms of PD [12, 142, 143]. Mutation of LRRK2 (R1441C) results in impaired autophagic balance and degradative capacity [142]. Alpha-synuclein inclusions, a component of Lewy bodies, are characteristic with dysfunctional autophagy [144]. Overexpression of α-synuclein, which is seen in forms of PD, impairs macroautophagy and increases p62 levels due to ATG9 mislocation [145]. PINK1 and Parkin are closely linked to mitochondria as mentioned above. PINK1 can recruit Parkin to the mitochondria, which mediate degradation of damaged mitochondria by autophagy. Mutations of PINK1 or Parkin abrogate this function [146]. Cells lacking ATP13A2 have impaired autophagosome-lysosome fusion [147]. GBA is the gene that encodes glucocerebrosidase, a lysosomal enzyme that degrades glucocerebroside. Homozygotes with GBA mutations typically develop a lysosomal storage disease called Gaucher’s disease. A number of published studies have found that heterozygosity for GBA mutations is associated with defects in autophagy and mitophagy as a consequence of impaired lysosomal activity [148, 149]. Thus, a parsimonious explanation for the PD risk associated with GBA heterozygosity is that it impairs lysosomal function and thereby compromises autophagic degradation of alpha-synuclein.
Previous studies support that oxidative stress and impairment of DNA repair pathways may contribute to PD [10, 150, 151]. PD patients exhibit polymorphisms in DNA repair genes, including XRCC1, XRCC3, and APE1, which are caused by oxidative stress [150]. Additionally, NER capacity may be impaired in PD patients, who have genetic mutations such as LRRK2 (G2019S) and (R1441G). Mutation of excision repair cross-complementation group1 (ERCC1), an essential factor in NER pathway increases dopaminergic (DA) neuron injury, loss of striatal DA neurons, DNA damage, inclusions of α-synuclein, and abnormal morphologic mitochondria in mice [10]. Similarly, ERCC1-mutated mice have early onset of age-related phenotypes, such as reduced autophagy and increased cellular senescence. Rapamycin treatment promotes autophagy and reduces cell senescence markers in ERCC1 mutant mice [152].
Amyotrophic lateral sclerosis
Patients with ALS show loss of motor neurons within the CNS and degeneration of neuromuscular junctions. Mutation of genes including SOD1, TAR DNA-binding protein 43 (TDP-43), OPTN, fused in sarcoma (FUS), p62, and chromosome 9 open reading frame 72 (C9ORF72), cause familial forms of ALS [12, 153]. Many of ALS-associated genes are involved in autophagy/lysosomal function [12]. An abnormal interaction between mutated form of SOD1 (G86R) and beclin1 may modulate autophagy. Haplosufficient beclin1 shows protective effects in mutant SOD1 transgenic mice [154]. On the other hand, in the early symptomatic stage of disease, mutant SOD1 (G93A) mice exhibit upregulation of beclin1 and TFEB; however, in the middle and end stages, these mice show decreased levels of beclin1 and TFEB. Overexpression of TFEB augments effect of the beclin1 [155]. Mutation of p62 (L341V) impairs its ability to recognize LC3B, thereby reducing p62 recruitment to phagophores [156]. Treatment of trehalose can reduce aggregates of SOD1 and p62, and induce autophagic flux by rescuing mitochondrial degeneration in mutant SOD1 (G93A) mice. Moreover, trehalose reduces oxidative stress in the muscle of SOD1 mutated mice [157]. In mutant SOD1 mice, trehalose can extend life span and retard progression of disease by enhancing autophagy-associated factors, such as LC3, p62, ATG5 and beclin1, although the amelioration of disease may also be due to chemical chaperone effects of this compound reducing mutant SOD1 aggregation [158]. Autophagy and the ubiquitin-proteasome system (UPS) are involved in TDP-43 removal: clearance of soluble TDP-43 by the UPS and insoluble TDP-43 by autophagy. ALS tissues exhibit abnormal cytoplasmic accumulation of TDP-43, which fails to be degraded by degradation adaptor proteins owing to its mutation or the adaptor’s mutation [159]. Autophagy inducers, including rapamycin, tamoxifen, carbamazepine, and spermidine, improve motor function and reduce the formation of TDP-43 inclusions in the brain of TDP-43 Tg mice. [160]. The most common mutations in ALS are hexanucleotide repeat expansions (GGGGCC) in C9ORF72. C9ORF72 is involved in the regulation of endosomal trafficking (RAB-dependent manner) for endosomal transport and autophagy. Deletion of C9ORF72 causes dysfunctional autophagy and endocytosis [161].
Age-related factors such as inflammatory responses and oxidative stress are related to the pathogenesis of ALS [11]. ALS-related genetic mutations result in inflammation [11]. Reactive microglia and macrophages are abundant in postmortem brain and spinal cord of ALS patients, and T lymphocyte infiltration is present in ALS tissue [162]. C9ORF72 deficient mice show morphological change in immune organs, such as spleen, liver, and lymph nides, and macrophage infiltration and increased levels of lysosomal proteins are observed in these mice [163].
In a mutant SOD1 (G93A) rat model of ALS, microgliosis and loss of motor neuron are observed in the spinal cord. In microglial primary cultures from symptomatic SOD1 (G93A) rats, senescence-associated bate-Galactosidase (SA-β-Gal), p53, and p16INK4a are increased during disease progression [164]. Mutant SOD1 (G93A) activates caspase-1-mediated IL-1β secretion in microglia and macrophages. Longevity is extended in mutant SOD1 (G93A) mice lacking caspase-1 or IL-1β. Autophagy inhibition results in increased SOD1 accumulation in the cytosolic compartment and released IL-1β levels. Under ATG5-deficient condition, caspase-1 activity is increased in response to mutant SOD1 (G93A) [165].
CONCLUDING REMARKS
In this review, we have briefly described possible connections between impaired autophagy and factors in cellular aging. Causative factors in aging, such as oxidative stress, DNA damage, telomere shortening, and SASP, are closely associated with autophagy. The interplay between aging and abnormal autophagy may accelerate age-related pathology and diseases (Table 1). Deficient autophagy may contribute to AD, PD, HD, and ALS, and contribute to the toxicity of aggregate-prone proteins. Under these disease conditions, pharmacological treatment for enhancing autophagy has been shown to ameliorate disease in various animal models [12, 166]. However, such strategies need to consider how modulation of autophagy may impact other aspects of neuronal and glial homeostasis [167]. Ultimately, understanding of autophagy mechanisms in the process of aging may provide crucial information for improving health and preventing disease states. Modulation of autophagy by pharmacological or genetic tools may be a potential clinical intervention for age-related diseases.
ACKNOWLEDGEMENTS
We are grateful for funding from the UK Dementia Research Institute (funded by the MRC, Alzheimer’s Research UK and the Alzheimer’s Society), Roger de Spoelberch Foundation, Alzheimer’s Research UK, The Tau Consortium, Cambridge Centre for Parkinson-Plus, National Institute for Health Research Cambridge Biomedical Research Centre (D.C.R.), and the Korea Health Technology R&D Project (HI14C2173) through the Korea Health Industry Development Institute, funded by the Ministry of Health & Welfare, Republic of Korea (S.Y.C., H. K., and J.E.L). The views expressed are those of the author(s) and not necessarily those of the NHS, the NIHR or the Department of Health and Social Care. The authors thank MID (Medical Illustration & Design), a part of the Medical Research Support Services of Yonsei University College of Medicine, for all artistic support related to this work.
Figures
{kind=link}
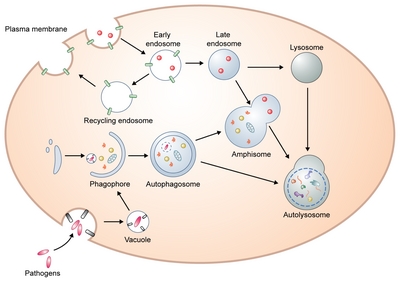
Schematic overview of the autophagy process.
Initially, a precursor form of the autophagosome, a phagophore is formed by various sources including ER, Golgi, mitochondria, endosomes, and plasma membrane. This cup-shaped and double membrane structure extends its edges to form an autophagosome. Closed autophagosomes containing engulfed cytoplasmic proteins or organelles migrate toward the perinuclear part of the cells where lysosomes are clustered to enable autophagosome-lysosome fusion. Alternatively, autophagosomes can generate amphisomes by fusion with endosomes. Ultimately, autophagosomes or amphisomes fuse with lysosomes where their contents are degraded.
Tables
Table 1 Impaired autophagy in age-related diseases
| Disease | Major causes and defects | References |
|---|---|---|
| Alzheimer’s disease | Clearance of misfolded plaque as autophagy substrate | [1, 111–113, 117–120] |
| Lysosomal. acidification | [112, 113, 116] | |
| Inflammatory cytokine & Reactive microglia | [8, 121–125, 127] | |
| Oxidative stress | [128–130] | |
| Mitochondrial oxidative damage | [131, 133] | |
| Huntington’s disease | Accumulation of mutant htt & Cell death | [134, 136–139] |
| Oxidative stress & DNA repair system | [9, 135, 137, 140, 141] | |
| Parkinson’s disease | Genetic mutations in autosomal dominant genes | [12, 142, 144, 145, 148, 149] |
| Genetic mutations in autosomal recessive genes | [12, 146, 147] | |
| Oxidative stress & DNA repair system | [10, 150–152] | |
| Amyotrophic lateral sclerosis | Mutation of SOD1 gene | [153–155, 157, 158] |
| Mutation of p62 | [153, 156] | |
| Abnormal cytoplasmic accumulation of TDP-43 | [153, 159, 160] | |
| Deletion of C9ORF72 | [161] | |
| Inflammatory responses & Reactive microglia | [11, 162–165] |
References
- Rubinsztein DC, Mariño G, Kroemer G (2011) Autophagy and aging. Cell 146:682-695.


- Hansen M, Rubinsztein DC, Walker DW (2018) Autophagy as a promoter of longevity: insights from model organisms. Nat Rev Mol Cell Biol 19:579-593.

- López-Otín C, Blasco MA, Partridge L, Serrano M, Kroemer G (2013) The hallmarks of aging. Cell 153:1194-1217.
- Balaban RS, Nemoto S, Finkel T (2005) Mitochondria, oxidants, and aging. Cell 120:483-495.
- Seluanov A, Mittelman D, Pereira-Smith OM, Wilson JH, Gorbunova V (2004) DNA end joining becomes less efficient and more error-prone during cellular senescence. Proc Natl Acad Sci U S A 101:7624-7629.
- Henriques CM, Ferreira MG (2012) Consequences of telomere shortening during lifespan. Curr Opin Cell Biol 24:804808.
- Sun Y, Coppé JP, Lam EW (2018) Cellular senescence: the sought or the unwanted? Trends Mol Med 24:871-885.
- Yao Y, Chinnici C, Tang H, Trojanowski JQ, Lee VM, Praticò D (2004) Brain inflammation and oxidative stress in a transgenic mouse model of Alzheimer-like brain amyloidosis. J Neuroinflammation 1:21.
- Budworth H, Harris FR, Williams P, Lee DY, Holt A, Pahnke J, Szczesny B, Acevedo-Torres K, Ayala-Peña S, McMurray CT (2015) Suppression of somatic expansion delays the onset of pathophysiology in a mouse model of Huntington's disease. PLoS Genet 11:e1005267.
- Sepe S, Milanese C, Gabriels S, Derks KW, Payan-Gomez C, van IJcken WF, Rijksen YM, Nigg AL, Moreno S, Cerri S, Blandini F, Hoeijmakers JH, Mastroberardino PG (2016) Inefficient DNA repair is an aging-related modifier of Parkinson's disease. Cell Rep 15:1866-1875.
- Beers DR, Appel SH (2019) Immune dysregulation in amyotrophic lateral sclerosis: mechanisms and emerging therapies. Lancet Neurol 18:211-220.
- Menzies FM, Fleming A, Caricasole A, Bento CF, Andrews SP, Ashkenazi A, Füllgrabe J, Jackson A, Jimenez Sanchez M, Karabiyik C, Licitra F, Lopez Ramirez A, Pavel M, Puri C, Renna M, Ricketts T, Schlotawa L, Vicinanza M, Won H, Zhu Y, Skidmore J, Rubinsztein DC (2017) Autophagy and neurodegeneration: pathogenic mechanisms and therapeutic opportunities. Neuron 93:1015-1034.
- Menzies FM, Fleming A, Rubinsztein DC (2015) Compromised autophagy and neurodegenerative diseases. Nat Rev Neurosci 16:345-357.
- Efeyan A, Zoncu R, Sabatini DM (2012) Amino acids and mTORC1: from lysosomes to disease. Trends Mol Med 18:524-533.
- Bernales S, Schuck S, Walter P (2007) ER-phagy: selective autophagy of the endoplasmic reticulum. Autophagy 3:285-287.
- Lipinski MM, Zheng B, Lu T, Yan Z, Py BF, Ng A, Xavier RJ, Li C, Yankner BA, Scherzer CR, Yuan J (2010) Genome-wide analysis reveals mechanisms modulating autophagy in normal brain aging and in Alzheimer's disease. Proc Natl Acad Sci U S A 107:14164-14169.
- Martinez-Lopez N, Athonvarangkul D, Singh R (2015) Autophagy and aging. Adv Exp Med Biol 847:73-87.
- Cuervo AM (2008) Autophagy and aging: keeping that old broom working. Trends Genet 24:604-612.
- Kim J, Kundu M, Viollet B, Guan KL (2011) AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat Cell Biol 13:132-141.
- Dooley HC, Razi M, Polson HE, Girardin SE, Wilson MI, Tooze SA (2014) WIPI2 links LC3 conjugation with PI3P, autophagosome formation, and pathogen clearance by recruiting Atg12-5-16L1. Mol Cell 55:238-252.
- Weidberg H, Shvets E, Shpilka T, Shimron F, Shinder V, Elazar Z (2010) LC3 and GATE-16/GABARAP subfamilies are both essential yet act differently in autophagosome biogenesis. EMBO J 29:1792-1802.
- Ravikumar B, Moreau K, Jahreiss L, Puri C, Rubinsztein DC (2010) Plasma membrane contributes to the formation of pre-autophagosomal structures. Nat Cell Biol 2010;12:747-757.
- Pavel M, Rubinsztein DC (2017) Mammalian autophagy and the plasma membrane. FEBS J 2017;284:672-679.
- Moreau K, Ravikumar B, Renna M, Puri C, Rubinsztein DC (2011) Autophagosome precursor maturation requires homotypic fusion. Cell 2011;146:303-317.
- Puri C, Renna M, Bento CF, Moreau K, Rubinsztein DC (2013) Diverse autophagosome membrane sources coalesce in recycling endosomes. Cell 154:1285-1299.
- Hamasaki M, Furuta N, Matsuda A, Nezu A, Yamamoto A, Fujita N, Oomori H, Noda T, Haraguchi T, Hiraoka Y, Amano A, Yoshimori T (2013) Autophagosomes form at ER-mitochondria contact sites. Nature 495:389-393.
- Puri C, Vicinanza M, Ashkenazi A, Gratian MJ, Zhang Q, Bento CF, Renna M, Menzies FM, Rubinsztein DC (2018) The RAB11A-positive compartment is a primary platform for autophagosome assembly mediated by WIPI2 recognition of PI3P-RAB11A. Dev Cell 45:114-131.e8.
- Ravikumar B, Acevedo-Arozena A, Imarisio S, Berger Z, Vacher C, O'Kane CJ, Brown SD, Rubinsztein DC (2005) Dynein mutations impair autophagic clearance of aggregateprone proteins. Nat Genet 37:771-776.
- Gross SP, Vershinin M, Shubeita GT (2007) Cargo transport:two motors are sometimes better than one. Curr Biol 17:R478-R486.
- Maday S, Wallace KE, Holzbaur EL (2012) Autophagosomes initiate distally and mature during transport toward the cell soma in primary neurons. J Cell Biol 196:407-417.
- Rubinsztein DC, Codogno P, Levine B (2012) Autophagy modulation as a potential therapeutic target for diverse diseases. Nat Rev Drug Discov 11:709-730.
- Hickey CM, Wickner W (2010) HOPS initiates vacuole docking by tethering membranes before trans-SNARE complex assembly. Mol Biol Cell 21:2297-2305.
- Zhao YG, Zhang H (2019) Autophagosome maturation: an epic journey from the ER to lysosomes. J Cell Biol 218:757770.
- Moreau K, Renna M, Rubinsztein DC (2013) Connections between SNAREs and autophagy. Trends Biochem Sci 38:5763.
- Finkel T, Holbrook NJ (2000) Oxidants, oxidative stress and the biology of ageing. Nature 408:239-247.
- Kadenbach B (2012) Introduction to mitochondrial oxidative phosphorylation. Adv Exp Med Biol 748:1-11.
- Wallace DC (2005) A mitochondrial paradigm of metabolic and degenerative diseases, aging, and cancer: a dawn for evolutionary medicine. Annu Rev Genet 39:359-407.
- Chouchani ET, Pell VR, James AM, Work LM, Saeb-Parsy K, Frezza C, Krieg T, Murphy MP (2016) A unifying mechanism for mitochondrial superoxide production during ischemiareperfusion injury. Cell Metab 23:254-263.
- St-Pierre J, Buckingham JA, Roebuck SJ, Brand MD (2002) Topology of superoxide production from different sites in the mitochondrial electron transport chain. J Biol Chem 277:44784-44790.
- Perier C, Vila M (2012) Mitochondrial biology and Parkinson's disease. Cold Spring Harb Perspect Med 2:a009332.
- Indo HP, Yen HC, Nakanishi I, Matsumoto K, Tamura M, Nagano Y, Matsui H, Gusev O, Cornette R, Okuda T, Minamiyama Y, Ichikawa H, Suenaga S, Oki M, Sato T, Ozawa T, Clair DK, Majima HJ (2015) A mitochondrial superoxide theory for oxidative stress diseases and aging. J Clin Biochem Nutr 56:1-7.
- Chen Y, Azad MB, Gibson SB (2009) Superoxide is the major reactive oxygen species regulating autophagy. Cell Death Differ 16:1040-1052.
- Shigenaga MK, Hagen TM, Ames BN (1994) Oxidative damage and mitochondrial decay in aging. Proc Natl Acad Sci U S A 91:10771-10778.
- Graef M, Nunnari J (2011) Mitochondria regulate autophagy by conserved signalling pathways. EMBO J 30:2101-2114.
- Lee J, Giordano S, Zhang J (2012) Autophagy, mitochondria and oxidative stress: cross-talk and redox signalling. Biochem J 441:523-540.
- Scherz-Shouval R, Elazar Z (2011) Regulation of autophagy by ROS: physiology and pathology. Trends Biochem Sci 36:30-38.
- Chen Y, Gibson SB (2008) Is mitochondrial generation of reactive oxygen species a trigger for autophagy? Autophagy 4:246-248.
- Li L, Chen Y, Gibson SB (2013) Starvation-induced autophagy is regulated by mitochondrial reactive oxygen species leading to AMPK activation. Cell Signal 25:50-65.
- Rabinovitch RC, Samborska B, Faubert B, Ma EH, Gravel SP, Andrzejewski S, Raissi TC, Pause A, St-Pierre J, Jones RG (2017) AMPK maintains cellular metabolic homeostasis through regulation of mitochondrial reactive oxygen species. Cell Rep 21:1-9.
- Xiao B, Deng X, Lim GGY, Xie S, Zhou ZD, Lim KL, Tan EK (2017) Superoxide drives progression of Parkin/PINK1dependent mitophagy following translocation of Parkin to mitochondria. Cell Death Dis 8:e3097.
- Khandelwal PJ, Herman AM, Hoe HS, Rebeck GW, Moussa CE (2011) Parkin mediates beclin-dependent autophagicclearance of defective mitochondria and ubiquitinated Abeta in AD models. Hum Mol Genet 20:2091-2102.
- Thomas KJ, McCoy MK, Blackinton J, Beilina A, van der Brug M, Sandebring A, Miller D, Maric D, Cedazo-Minguez A, Cookson MR (2011) DJ-1 acts in parallel to the PINK1/parkin pathway to control mitochondrial function and autophagy. Hum Mol Genet 20:40-50.
- Gautier CA, Kitada T, Shen J (2008) Loss of PINK1 causes mitochondrial functional defects and increased sensitivity to oxidative stress. Proc Natl Acad Sci U S A 105:11364-11369.
- Meulener MC, Xu K, Thomson L, Ischiropoulos H, Bonini NM (2006) Mutational analysis of DJ-1 in Drosophila implicates functional inactivation by oxidative damage and aging. Proc Natl Acad Sci U S A 103:12517-12522.
- Hao LY, Giasson BI, Bonini NM (2010) DJ-1 is critical for mitochondrial function and rescues PINK1 loss of function. Proc Natl Acad Sci U S A 107:9747-9752.
- Sirbu BM, Cortez D (2013) DNA damage response: three levels of DNA repair regulation. Cold Spring Harb Perspect Biol 5:a012724.
- Chen JH, Hales CN, Ozanne SE (2007) DNA damage, cellular senescence and organismal ageing: causal or correlative? Nucleic Acids Res 35:7417-7428.
- Fleck O, Nielsen O (2004) DNA repair. J Cell Sci 117:515-517.
- Maher LJ 3rd (2005) Indirect detection of DNA damage. Chem Biol 12:862-864.
- Hoeijmakers JH (2009) DNA damage, aging, and cancer. N Engl J Med 361:1475-1485.
- Swain U, Rao KS (2012) Age-dependent decline of DNA base excision repair activity in rat cortical neurons. Mech Ageing Dev 133:186-194.
- Goukassian D, Gad F, Yaar M, Eller MS, Nehal US, Gilchrest BA (2000) Mechanisms and implications of the age-associated decrease in DNA repair capacity. FASEB J 2000;14:13251334.
- Hewitt G, Korolchuk VI (2017) Repair, reuse, recycle: the expanding role of autophagy in genome maintenance. Trends Cell Biol 27:340-351.
- Karantza-Wadsworth V, Patel S, Kravchuk O, Chen G, Mathew R, Jin S, White E (2007) Autophagy mitigates metabolic stress and genome damage in mammary tumorigenesis. Genes Dev 21:1621-1635.
- Wang Y, Zhang N, Zhang L, Li R, Fu W, Ma K, Li X, Wang L, Wang J, Zhang H, Gu W, Zhu WG, Zhao Y (2016) Autophagy regulates chromatin ubiquitination in DNA damage response through elimination of SQSTM1/p62. Mol Cell 63:34-48.
- Mathew R, Karp CM, Beaudoin B, Vuong N, Chen G, Chen HY, Bray K, Reddy A, Bhanot G, Gelinas C, Dipaola RS, Karantza-Wadsworth V, White E (2009) Autophagy suppresses tumorigenesis through elimination of p62. Cell 137:10621075.
- Hewitt G, Carroll B, Sarallah R, Correia-Melo C, Ogrodnik M, Nelson G, Otten EG, Manni D, Antrobus R, Morgan BA, von Zglinicki T, Jurk D, Seluanov A, Gorbunova V, Johansen T, Passos JF, Korolchuk VI (2016) SQSTM1/p62 mediates crosstalk between autophagy and the UPS in DNA repair. Autophagy 12:1917-1930.
- Gillespie DA, Ryan KM (2015) Autophagy is critically required for DNA repair by homologous recombination. Mol Cell Oncol 3:e1030538.
- Qiang L, Zhao B, Shah P, Sample A, Yang S, He YY (2016) Autophagy positively regulates DNA damage recognition by nucleotide excision repair. Autophagy 12:357-368.
- Mathew R, Kongara S, Beaudoin B, Karp CM, Bray K, Degenhardt K, Chen G, Jin S, White E (2007) Autophagy suppresses tumor progression by limiting chromosomal instability. Genes Dev 21:1367-1381.
- Maynard S, Schurman SH, Harboe C, de Souza-Pinto NC, Bohr VA (2009) Base excision repair of oxidative DNA damage and association with cancer and aging. Carcinogenesis 30:2-10.
- Du Y, Wooten MC, Wooten MW (2009) Oxidative damage to the promoter region of SQSTM1/p62 is common to neurodegenerative disease. Neurobiol Dis 35:302-310.
- Kauppila TES, Kauppila JHK, Larsson NG (2017) Mammalian mitochondria and aging: an update. Cell Metab 25:57-71.
- Muftuoglu M, Mori MP, de Souza-Pinto NC (2014) Formation and repair of oxidative damage in the mitochondrial DNA. Mitochondrion 17:164-181.
- Zeng X, Kinsella TJ (2010) BNIP3 is essential for mediating 6-thioguanine- and 5-fluorouracil-induced autophagy following DNA mismatch repair processing. Cell Res 20:665675.
- Zhang J, Ney PA (2009) Role of BNIP3 and NIX in cell death, autophagy, and mitophagy. Cell Death Differ 16:939-946.
- Eitan E, Hutchison ER, Mattson MP (2014) Telomere shortening in neurological disorders: an abundance of unanswered questions. Trends Neurosci 7:256-263.
- de Lange T (2005) Shelterin: the protein complex that shapes and safeguards human telomeres. Genes Dev 9:2100-2110.
- Boccardi V, Pelini L, Ercolani S, Ruggiero C, Mecocci P (2015) From cellular senescence to Alzheimer's disease: the role of telomere shortening. Ageing Res Rev 22:1-8.
- Rudolph KL, Chang S, Lee HW, Blasco M, Gottlieb GJ, Greider C, DePinho RA (1999) Longevity, stress response, and cancer in aging telomerase-deficient mice. Cell 96:701-712.
- Samper E, Flores JM, Blasco MA (2001) Restoration of telomerase activity rescues chromosomal instability and premature aging in Terc-/- mice with short telomeres. EMBO Rep 2:800807.
- Counter CM, Avilion AA, LeFeuvre CE, Stewart NG, Greider CW, Harley CB, Bacchetti S (1992) Telomere shortening associated with chromosome instability is arrested in immortal cells which express telomerase activity. EMBO J 11:19211929.
- Hande MP, Samper E, Lansdorp P, Blasco MA (1999) Telomere length dynamics and chromosomal instability in cells derived from telomerase null mice. J Cell Biol 144:589-601.
- Blasco MA, Lee HW, Hande MP, Samper E, Lansdorp PM, DePinho RA, Greider CW (1997) Telomere shortening and tumor formation by mouse cells lacking telomerase RNA. Cell 91:25-34.
- Vaziri H, West MD, Allsopp RC, Davison TS, Wu YS, Arrowsmith CH, Poirier GG, Benchimol S (1997) ATM-dependent telomere loss in aging human diploid fibroblasts and DNA damage lead to the post-translational activation of p53 protein involving poly(ADP-ribose) polymerase. EMBO J 16:6018-6033.
- Chin L, Artandi SE, Shen Q, Tam A, Lee SL, Gottlieb GJ, Greider CW, DePinho RA (1999) p53 deficiency rescues the adverse effects of telomere loss and cooperates with telomere dysfunction to accelerate carcinogenesis. Cell 97:527-538.
- Aoki H, Iwado E, Eller MS, Kondo Y, Fujiwara K, Li GZ, Hess KR, Siwak DR, Sawaya R, Mills GB, Gilchrest BA, Kondo S (2007) Telomere 3' overhang-specific DNA oligonucleotides induce autophagy in malignant glioma cells. FASEB J 21:2918-2930.
- Nassour J, Radford R, Correia A, Fusté JM, Schoell B, Jauch A, Shaw RJ, Karlseder J (2019) Autophagic cell death restricts chromosomal instability during replicative crisis. Nature 565:659-663.
- Ali M, Devkota S, Roh JI, Lee J, Lee HW (2016) Telomerase reverse transcriptase induces basal and amino acid starvationinduced autophagy through mTORC1. Biochem Biophys Res Commun 478:1198-1204.
- White E (2016) Autophagy and p53. Cold Spring Harb Perspect Med 6:a026120.