Articles

Article Tools
 View Full Text View Full Text |
 Abstract Abstract |
 Article as PDF Article as PDF |
 Print this Article Print this Article |
 Pubmed Pubmed |
 PMC PMC |
 PubReader PubReader |
 Export to Citation Export to Citation |
 Email Alerts Email Alerts |
 Open Access Open Access |
Share this article on :



Stats or Metrics
Article
Original Article
Exp Neurobiol 2013; 22(3): 200-207
Published online September 30, 2013
https://doi.org/10.5607/en.2013.22.3.200
© The Korean Society for Brain and Neural Sciences
4-hydroxy-2(E)-Nonenal facilitates NMDA-Induced Neurotoxicity via Triggering Mitochondrial Permeability Transition Pore Opening and Mitochondrial Calcium Overload
In-Young Choi1†, Ji-Hyae Lim2†, Chunsook Kim3, Hwa Young Song1, Chung Ju1 and Won-Ki Kim1*
1Department of Neuroscience, College of Medicine, Korea University, Seoul 136-705, 2Laboratory of Medical Genetics, Medical Research Institute, Cheil General Hospital and Women's Healthcare Center, Seoul 100-280, 3Department of Nursing, Andong Science College, Andong 760-709, Korea
Correspondence to: *To whom correspondence should be addressed.
TEL: 82-2-920-6094, FAX: 82-2-953-6095
e-mail: wonki@korea.ac.kr
†Equal contribution for the present study
Keywords: 4-hydroxy-2(E)-nonenal (HNE), NMDA, neuronal death, calcium, mitochondria
INTRODUCTION
In cerebral ischemia/reperfusion, neuronal cell injury or death is mediated by multiplex factors such as massive accumulation of extracellular glutamate, intracellular calcium overload, free radicals-induced oxidative stress, mitochondrial dysfunction, inflammatory reaction, and others. In general, excess release of glutamate causes over-stimulation of its receptors, including NMDA and AMPA/kainate receptor subtypes, and sustained increase of intracellular calcium level. Intracellular calcium overload in turn induces oxidative stress and damages the cellular membrane and microorganelles including mitochondria [1]. Even though each of these deteriorating events is accepted as one of the key factors for neuronal death, the interplay among the events has also been suggested to be important for neuronal cell death [2].
Mitochondria play a crucial role in the regulation of intracellular calcium concentration ([Ca2+]i), oxidant level, and ATP synthesis. Thus, mitochondrial damage is recognized as a common neuropathogenesis in numerous brain diseases, including Alzheimer's disease, Parkinson's disease, and cerebral ischemia/reperfusion [3, 4]. Prolonged oxidative stimuli cause mitochondrial dysfunction, including the decrease of mitochondrial transmembrane potential (ΔΨm), overload of mitochondrial calcium, and opening of mitochondrial permeability transition pore (MPTP) [5, 6].
Lipid peroxidation by oxidative stresses does much harm to neurons. Lipid peroxidation is an important mode of oxidative damage, particularly in the brain that is enriched with lipid molecules such as polyunsaturated fatty acids. A major deleterious outcome of lipid peroxidation is the generation of reactive aldehydes, and the most cytotoxic product among those is 4-hydroxy-2(
HNE has earlier been shown to increase the vulnerability of neurons to glutamate receptor-mediated excitotoxicity [9, 16]. We also reported that HNE induced synergistic death of neurons when treated together with NMDA [11]. However, the exact mechanism for the synergistic cytotoxicity by HNE and NMDA remains largely unclear. In the present study, we demonstrated for the first time that the rise of [Ca2+]i by NMDA facilitates HNE-induced mitochondrial calcium overload and dysfunction, and eventually neuronal cell death.
MATERIALS AND METHODS
Primary cultures of rat cortical neurons were prepared from embryonic 16 days old fetal Sprague-Dawley rats. In brief, brain tissues free of meninges were dissociated by triturating through a Pasteur pipette, and cell suspension (cell number: 5×105 cells/ml) was added onto the culture plates pre-coated with poly-D-lysine and laminin. Cells were maintained in Dulbecco's modified Eagle's medium (DMEM) containing 10% heat-inactivated fetal bovine serum (FBS) in humidified 95% air/5% CO2 at 37℃. Three days later, cytosine arabinoside (5 µM) was added for 1 day to block non-neuronal cell division. Subsequently medium was replaced twice a week. Experiments were performed on 16-18 days cultures.
Cells were exposed to HNE (1 µM) and/or NMDA (2 µM) in nominally Mg2+-free Hank's balanced salt solution (HBSS) containing 1.5 mM CaCl2, 1 mM glycine, and 10 mM glucose. For the calcium-free condition, CaCl2 was not included in the HBSS. Six hours later, cell injury or death was assessed by the morphological examination using a phase-contrast microscopy and quantified by measuring the amount of lactate dehydrogenase (LDH) released into the bathing medium. LDH activity was measured using a diagnostic kit (Sigma Chemical Co., St. Louis, MO). Cell death was expressed as percentage of total LDH, which was measured in sister cultures frozen and thawed after the experiments.
[Ca2+]i was measured fluorometrically according to the fura-2 method, as we described before (Lim et al., 2006). Cells were cultured on poly-D-lysine and laminin-coated coverslips (diameter=13 mm). They were then washed twice with HBSS, incubated with 1 µM fura-2AM for 30 min at 37℃, and then washed twice with HBSS. A coverslip with cells was diagonally placed inside the cuvette, and the bathing medium was stirred with a teflon-coated magnetic bar to allow adequate mixing of added drugs. The fluorescence was stimulated at 340 and 380 nm of incident light and collected at 510 nm. Constants were determined for each coverslip with 0.1% Triton-X100 for Rmax and 40 mM EGTA for Rmin. The ratios of [Ca2+]i at 340 an 380 nm were calculated according to the formula: [Ca2+]i=
Cells were loaded with JC-1 (1.0 µg/ml; Molecular Probes, Eugene, OR) for 20 min at 37℃. Depolarization of ΔΨm was assessed by measuring fluorescence intensities at the excitation wavelength of 485 nm and dual emission wavelengths of 530 and 590 nm (for measuring JC-1 monomers and aggregates, respectively) using a fluorescence microplate reader (SpectraMax GeminiXS, Molecular Devices, CA). During measurements, cells were maintained at 37℃ and protected from light. Fluorescence intensity was measured every 15 or 30 min. All fluorescent measurements were corrected by autofluorescence that was determined in cells not loaded with JC-1. Autofluorescence was constant throughout the experiment. In control experiments, no photobleaching was observed during fluorescence monitoring.
In order to measure [Ca2+]m, cultured cortical neurons were incubated with the mitochondrial Ca2+-sensitive fluorescent indicator, rhod-2 AM (5 µM, Molecular Probes), for 30 min at room temperature. Rhod-2 fluorescence was excited at 560 nm and emitted fluorescence was collected through a 590 nm long pass barrier filter using a fluorescence microplate reader (SpectraMax GeminiXS, Molecular Devices, CA). The rhod-2 fluorescence intensities were expressed as F/F0 (F0: fluorescence before drug addition, F: maximal fluorescence after drug addition).
Mitochondria were isolated from livers of male Sprague-Dawley rats (220-280 g) (Charles River Breeding Lab), as we described before [17]. Rats were starved overnight before sacrifice. All steps of mitochondria isolation were carried out on ice and in a cold medium containing 0.25 M sucrose, 2 mM K+-EDTA, and 3 mM HEPES, adjusted to pH 7.4 with KOH. Rat liver was washed, minced, and homogenized on ice using a ground-glass homogenizer. The homogenate was subjected to centrifugation at 600 g for 10 min, and the supernatant obtained to 8,000 g for 20 min. The subsequent mitochondrial pellet was washed in EDTA-free medium and used for experiments. Mitochondrial protein concentration was determined by the Bradford method using bovine serum albumin as the standard.
The ΔΨm in isolated mitochondria was measured using JC-1 [17] or rhodamin 123 [18]. Rhodamine 123 has spectral and metabolic properties as a probe of transmembrane potential in isolated rat-liver mitochondria. Thus, mitochondria (0.5 mg protein) were suspended with 2 ml of recording buffer containing 150 mM sucrose, 5 mM MgCl2, 5 mM succinate, 2.7 µM rotenone, 200 nM Ca2+, 50 mM potassium phosphate, and 20 mM HEPES (pH 7.4). Before each experiment, mitochondria in the recording medium were incubated with each dye [0.2 µM rhodamine 123 (Molecular Probes) or JC-1 (1.0 µg/ml)], for 20 min in the dark in a 3 ml recording cuvette at 35℃. The emission signals at 590 and 530 nm elicited by the excitation at 485 nm for JC-1 and the emission signals at 529 nm elicited by excitation at 507 nm for rhodamin 123 were measured with a spectrofluorophotometer RF-5301PC (Shimadzu, Kyoto, Japan). The ratio of the signal at 590 nm over that at 530 nm (red/green ratio) of JC-1 or fluorescence intensity of rhodamin 123 was calculated to estimate ΔΨm. The recording chamber was magnetically stirred, and the measurements were carried out at 35℃.
MPTP opening was assessed by measuring mitochondrial swelling under energized and de-energized conditions, as described previously [19]. Mitochondrial swelling was determined spectrophotometrically (Shimadzu UV-2401PC, Kyoto, Japan) by measuring absorbance changes at 540 nm. Mitochondria (1 mg protein) were incubated at 25℃ under energized conditions in 2 ml of medium containing 13 mM mannitol, 70 mM sucrose, 3 mM HEPES (pH 7.4), 10 mM succinate, and 1 µM rotenone. When required, de-energized condition was achieved by incubating mitochondria in KSCN buffer (150 mM KSCN, 20 mM MOPS, 10 mM Tris, 0.5 µM rotenone, and 0.5 mM antimycin, pH 7.4).
The level of intracellular ATP was measured using the method described previously (Choi et al., 2002). Briefly, cells were lysed with 10% trichloroacetic acid, sonicated for 1 min on ice, and 2 mM EDTA and 2 mg/ml bovine serum albumin (BSA) were added with. After centrifugation, the supernatant was collected and neutralized with 4 N to the lysate. The ATP content was determined using a luminescence detection kit (Molecular Probes, Eugene, OR).
Data are expressed as mean ± standard error of mean (S.E.M.) and analysed for statistical significance using repeated measures of ANOVA or two-way ANOVA by running SAS Window v.9.1. program. If needed, post-hoc Scheffe's test was performed for multiple comparisons. A p value <0.05 was considered significant.
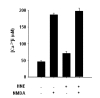
In the present study, neither HNE (1 µM) nor NMDA (2 µM) alone injured neuronal cells (Fig. 1A). However, combined treatment of the cells with HNE and NMDA evoked the release of LDH (Fig. 1A) with apparent morphological deterioration (data not shown). Furthermore, it is of a great interest to note that the synergistic death was observed only in the presence of calcium (Fig. 1). HNE alone neither increased the cytosolic calcium level ([Ca2+]i) nor altered the NMDA-induced intracellular calcium influx (Fig. 2), which is consistent with our previous observation (Lim et al., 2006). Therefore, our previous and present results together suggest that NMDA-mediated [Ca2+]i rise interplays with HNE to augment the neuronal cell death.
Mitochondrial dysfunction is one of the prominent features of oxidative stress-mediated cell death. Oxidative stress, including HNE, has been shown to depolarize the ΔΨm and facilitate Ca2+-evoked MPTP opening [20]. Similarly, NMDA receptor activation is also known to induce mitochondrial dysfunction [21]. As shown in Fig. 3A, however, neither 1 µM HNE nor 2 µM NMDA altered the ΔΨm in neuronal cells (Fig. 3A). On the other hand, when the cells were incubated for 3 h with 1 µM HNE together with 2 µM NMDA, there was complete depolarization of ΔΨm (Fig. 3A), which was comparable to the extent induced by 5 µM FCCP, a potent mitochondrial uncoupler (data not shown). Interestingly, the potentiated depolarization of ΔΨm by HNE and NMDA occurred only in the presence of extracellular calcium (Fig. 3A, B). Accumulation of calcium in mitochondria is well associated with the depolarization and dysfunction of mitochondria. Our results clearly showed that co-treatment with HNE and NMDA increased the [Ca2+]m in cortical neurons (Fig. 3C). However, neither 1 µM HNE nor 2 µM NMDA alone showed any significant changes in [Ca2+]m (Fig. 3C). The results imply that the synergistic cytotoxicity by HNE and NMDA may be related to the rise of [Ca2+]m and mitochondrial dysfunction.
Our previous and present data showed that 2 µM NMDA elevated the [Ca2+]i up to 200 nM. Therefore, we tested in isolated mitochondria whether HNE (1 µM) could induce ΔΨm depolarization and MPTP opening in the presence of 200 nM calcium. Neither 1 µM HNE nor 200 nM calcium alone affected ΔΨm in isolated mitochondria (data not shown). However, 1 µM HNE significantly depolarized ΔΨm in the presence of 200 nM calcium in the test buffer (Fig. 4). Mitochondrial swelling experiments also showed that 1 µM HNE strongly promoted the MPTP opening induced by 200 nM CaCl2 in both energized (Fig. 5A) and de-energized (Fig. 5B) conditions. These findings indicate that HNE facilitates the calcium-induced opening of MPTP. The HNE/Ca2+-facilitated MPTP opening shown in Fig. 4 was completely blocked by two different MPTP blockers, cyclosporine A and ciclopirox (data not shown).
We previously reported that ciclopirox protected adenocarcinoma cells from H2O2 toxicity via complete prevention of ΔΨm decrease and MPTP opening [17]. Unlike cell membrane-impermeable cyclosporine A or bonkrekic acid, the cell membrane-permeable MPTP blocker ciclopirox rapidly and markedly reduced mitochondrial damage induced by H2O2 in cultured cells. In the present study, therefore, we further examined whether ciclopirox blocked the synergistic toxicity of HNE and NMDA on mitochondria, and found that combined treatment of HNE and NMDA strongly increased the [Ca2+]m (Fig. 6A) and synergistically decreased the intracellular ATP level (Fig. 6B). However, ciclopirox completely blocked the mitochondrial calcium overload, restored intracellular ATP levels and ameliorated the neuronal cell death caused by HNE and NMDA (Fig. 6).
Microdialysis study showed that the level of glutamate in cerebral ischemic lesion reached over 70 µM, whereas HNE is normally present in human plasma below 0.1 µM, but it can reach up to 1 mM in response to oxidative insults [8]. Therefore, the concentrations of HNE (1 µM) and NMDA (2 µM) used in this study should be pathologically relevant. At the concentrations used in this study, treatment with HNE or NMDA alone did not evoke neuronal cell injury/death, however, combined treatment with HNE and NMDA together markedly increased the neuronal cell death, implying that concerted play of NMDA and HNE could be responsible for the massive brain damage during the early period of ischemic insult.
One possible mechanism for neurotoxicity of HNE has been suggested as the consequence of intracellular calcium overload [9, 20, 21]. This possibility is supported by the previous findings that HNE induces the impairment of ion transporters, voltage-dependent Ca2+ channels and glucose or glutamate transporters, and the phosphorylation of NMDA receptor subunit, resulting in intracellular calcium overload [20, 21]. However, the intracellular calcium overload was observed only at concentrations of HNE higher than that used in the present study: HNE significantly increased the [Ca2+]i at >10-fold higher concentrations in PC12 cells, hippocampal neurons, and spinal cord neurons [9, 20]. Furthermore, the [Ca2+]i rise by HNE may be dependent on the cell type. For example, HNE at very high concentrations (25 or 440 µM) failed to increase [Ca2+]i in human platelets [22], whereas 1 µM HNE increased [Ca2+]i in hepatocytes [23]. In our present and previous studies [11], 1 µM HNE did not induce any changes in [Ca2+]i in control or NMDA-treated neurons.
Even though NMDA-evoked [Ca2+]i rise was not changed by HNE, our present results showed that the [Ca2+]i enhanced by NMDA plays a crucial role in the neuronal cell death in the presence of HNE, because the synergistic cell death by co-treatment of HNE and NMDA was completely attenuated by a specific NMDA receptor antagonist MK-801 [11], or by removing extracellular calcium (the present study). A question arises, therefore, how HNE potentiates NMDA-induced neuronal death with no alteration of intracellular calcium level. One of key mechanisms for HNE cytotoxicity is mitochondrial failure via mitochondrial calcium overload [24, 25], and NMDA neurotoxicity has also been suggested to be caused by the mitochondrial calcium accumulation [26, 27]. At concentrations used in this study, however, neither HNE nor NMDA alone altered the [Ca2+]m (the present results), but combined together, they evoked Ca2+ accumulation in mitochondria. Previously, HNE has been reported to induce the opening of MPTP in isolated fetal brain mitochondria [28]. HNE enhances calcium-mediated MPTP opening via permeabilizing adenine nucleotide translocator that is a component of MPTP and has been suggested as the only target of HNE [29, 30]. Furthermore, HNE inhibits efflux of Ca2+ from mitochondria by inhibiting pyridine nucleotide hydrolysis [31]. In the present study, we found that 2 µM HNE evoked MPTP opening in isolated mitochondria at very low calcium level (200 nM) induced by 1 µM NMDA. This synergistic calcium accumulation in mitochondria and opening of MPTP may result in the failure of mitochondrial ATP generation, as shown in Fig. 6B of this study.
Much effort has been paid to develop drugs for the treatment of oxidative stress-mediated neurodegenerative diseases. Mitochondrial damage is a common phenomenon in the pathogenesis of numerous brain diseases, including Alzheimer's disease, Parkinson's disease, and cerebral ischemic stroke [3, 4, 32]. We previously demonstrated that ciclopirox markedly reduced peroxynitrite- or hydrogen peroxide-induced mitochondrial damage and cell death by inhibiting ΔΨm decrease and MPTP opening [17, 33]. In the present study, we also observed that ciclopirox blocked the HNE plus NMDA-induced toxic effects on mitochondria. Therefore, further studies are needed in view of medicinal chemistry to develop new therapeutic drugs for protection of mitochondrial damage and resulting cell death.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
- Kristián T, Siesjö BK. Calcium in ischemic cell death. Stroke 1998;29:705-718.

- Chinopoulos C, Adam-Vizi V. Calcium, mitochondria and oxidative stress in neuronal pathology. Novel aspects of an enduring theme. FEBS J 2006;273:433-450.
- Sims NR, Anderson MF. Mitochondrial contributions to tissue damage in stroke. Neurochem Int 2002;40:511-526.
- Abou-Sleiman PM, Muqit MM, Wood NW. Expanding insights of mitochondrial dysfunction in Parkinson's disease. Nat Rev Neurosci 2006;7:207-219.
- Duchen MR. Mitochondria and Ca2+ in cell physiology and pathophysiology. Cell Calcium 2000;28:339-348.
- Abramov AY, Canevari L, Duchen MR. β-amyloid peptides induce mitochondrial dysfunction and oxidative stress in astrocytes and death of neurons through activation of NADPH oxidase. J Neurosci 2004;24:565-575.
- Comporti M. Lipid peroxidation and biogenic aldehydes: from the identification of 4-hydroxynonenal to further achievements in biopathology. Free Radic Res 1998;28:623-635.
- Esterbauer H, Schaur RJ, Zollner H. Chemistry and biochemistry of 4-hydroxynonenal, malonaldehyde and related aldehydes. Free Radic Biol Med 1991;11:81-128.
- Mark RJ, Lovell MA, Markesbery WR, Uchida K, Mattson MP. A role for 4-hydroxynonenal, an aldehydic product of lipid peroxidation, in disruption of ion homeostasis and neuronal death induced by amyloid β-peptide. J Neurochem 1997;68:255-264.
- McKracken E, Graham DI, Nilsen M, Stewart J, Nicoll JA, Horsburgh K. 4-hydroxynonenal immunoreactivity is increased in human hippocampus after global ischemia. Brain Pathol 2001;11:414-421.
- Lim JH, Lee JC, Lee YH, Choi IY, Oh YK, Kim HS, Park JS, Kim WK. Simvastatin prevents oxygen and glucose deprivation/reoxygenation-induced death of cortical neurons by reducing the production and toxicity of 4-hydroxy-2E-nonenal. J Neurochem 2006;97:140-150.
- Yabuki Y, Fukunaga K. Oral administration of glutathione improves memory deficits following transient brain ischemia by reducing brain oxidative stress. Neuroscience 2013;250C:394-407.
- Wang R, Tu J, Zhang Q, Zhang X, Zhu Y, Ma W, Cheng C, Brann DW, Yang F. Genistein attenuates ischemic oxidative damage and behavioral deficits via eNOS/Nrf2/HO-1 signaling. Hippocampus 2013;23:634-647.
- Guo C, Tong L, Xi M, Yang H, Dong H, Wen A. Neuroprotective effect of calycosin on cerebral ischemia and reperfusion injury in rats. J Ethnopharmacol 2012;144:768-774.
- Lee WC, Wong HY, Chai YY, Shi CW, Amino N, Kikuchi S, Huang SH. Lipid peroxidation dysregulation in ischemic stroke: plasma 4-HNE as a potential biomarker?. Biochem Biophys Res Commun 2012;425:842-847.
- Keller JN, Hanni KB, Markesbery WR. 4-hydroxynonenal increases neuronal susceptibility to oxidative stress. J Neurosci Res 1999;58:823-830.
- Lee SJ, Jin Y, Yoon HY, Choi BO, Kim HC, Oh YK, Kim HS, Kim WK. Ciclopirox protects mitochondria from hydrogen peroxide toxicity. Br J Pharmacol 2005;145:469-476.
- Emaus RK, Grunwald R, Lemasters JJ. Rhodamine 123 as a probe of transmembrane potential in isolated rat-liver mitochondria: spectral and metabolic properties. Biochim Biophys Acta 1986;850:436-448.
- Halestrap AP, Woodfield KY, Connern CP. Oxidative stress, thiol reagents, and membrane potential modulate the mitochondrial permeability transition by affecting nucleotide binding to the adenine nucleotide translocase. J Biol Chem 1997;272:3346-3354.
- Kruman I, Bruce-Keller AJ, Bredesen D, Waeg G, Mattson MP. Evidence that 4-hydroxynonenal mediates oxidative stress-induced neuronal apoptosis. J Neurosci 1997;17:5089-5100.
- Lu C, Chan SL, Fu W, Mattson MP. The lipid peroxidation product 4-hydroxynonenal facilitates opening of voltage-dependent Ca2+ channels in neurons by increasing protein tyrosine phosphorylation. J Biol Chem 2002;277:24368-24375.
- Fowler CJ, Ando Y, Tiger G. Comparison of the effects of hydrogen peroxide, 4-hydroxy-2-nonenal and β-amyloid (25-35) upon calcium signalling. Neurochem Int 1998;33:161-172.
- Carini R, Bellomo G, Paradisi L, Dianzani MU, Albano E. 4-hydroxynonenal triggers Ca2+ influx in isolated rat hepatocytes. Biochem Biophys Res Commun 1996;218:772-776.
- Kruman II, Mattson MP. Pivotal role of mitochondrial calcium uptake in neural cell apoptosis and necrosis. J Neurochem 1999;72:529-540.
- Camandola S, Poli G, Mattson MP. The lipid peroxidation product 4-hydroxy-2,3-nonenal increases AP-1-binding activity through caspase activation in neurons. J Neurochem 2000;74:159-168.
- Pivovarova NB, Stanika RI, Watts CA, Brantner CA, Smith CL, Andrews SB. Reduced calcium-dependent mitochondrial damage underlies the reduced vulnerability of excitotoxicity-tolerant hippocampal neurons. J Neurochem 2008;104:1686-1699.
- Fernandes HB, Baimbridge KG, Church J, Hayden MR, Raymond LA. Mitochondrial sensitivity and altered calcium handling underlie enhanced NMDA-induced apoptosis in YAC128 model of Huntington's disease. J Neurosci 2007;27:13614-13623.
- Ramachandran V, Perez A, Chen J, Senthil D, Schenker S, Henderson GI. In utero ethanol exposure causes mitochondrial dysfunction, which can result in apoptotic cell death in fetal brain: a potential role for 4-hydroxynonenal. Alcohol Clin Exp Res 2001;25:862-871.
- Kristal BS, Park BK, Yu BP. 4-hydroxyhexenal is a potent inducer of the mitochondrial permeability transition. J Biol Chem 1996;271:6033-6038.
- Yan LJ, Sohal RS. Mitochondrial adenine nucleotide translocase is modified oxidatively during aging. Proc Natl Acad Sci U S A 1998;95:12896-12901.
- Richter C, Meier P. Inhibition of pro-oxidant-induced mitochondrial pyridine nucleotide hydrolysis and calcium release by 4-hydroxynonenal. Biochem J 1990;269:735-737.
- Takuma K, Yan SS, Stern DM, Yamada K. Mitochondrial dysfunction, endoplasmic reticulum stress, and apoptosis in Alzheimer's disease. J Pharmacol Sci 2005;97:312-316.
- Choi JJ, Kong MY, Lee SJ, Kim HC, Ko KH, Kim WK. Ciclopirox prevents peroxynitrite toxicity in astrocytes by maintaining their mitochondrial function: a novel mechanism for cytoprotection by ciclopirox. Neuropharmacology 2002;43:408-417.