Articles

Article Tools
 View Full Text View Full Text |
 Abstract Abstract |
 Article as PDF Article as PDF |
 Print this Article Print this Article |
 Pubmed Pubmed |
 PMC PMC |
 PubReader PubReader |
 Export to Citation Export to Citation |
 Email Alerts Email Alerts |
 Open Access Open Access |
Share this article on :



Stats or Metrics
Article
Original Article
Exp Neurobiol 2018; 27(3): 171-180
Published online June 30, 2018
https://doi.org/10.5607/en.2018.27.3.171
© The Korean Society for Brain and Neural Sciences
LRRK2 Kinase Activity Induces Mitochondrial Fission in Microglia via Drp1 and Modulates Neuroinflammation
Dong Hwan Ho1†, A Reum Je2†, Haejin Lee2, Ilhong Son1,3, Hee-Seok Kweon2*,Hyung-Gun Kim4* and Wongi Seol1*
1InAm Neuroscience Research Center, Sanbon Medical Center, College of Medicine, Wonkwang University, Gunpo 15865, 2Electron Microscopy Research Center, Korea Basic Science Institute (KBSI), Daejeon 34133, 3Department of Neurology, Sanbon Medical Center, College of Medicine, Wonkwang University, Gunpo 15865, 4Department of Pharmacology, College of Medicine, Dankook University, Cheonan 31116, Korea
Correspondence to: *To whom correspondence should be addressed.
Hee-Seok Kweon, TEL: 82-43-240-5440, FAX: 82-42-865-3939
e-mail: hskweon@kbsi.re.kr
Hyung-Gun Kim, TEL: 82-41-550-3867, FAX: 82-41-551-3866
e-mail: hgkimm@dankook.ac.kr
Wongi Seol, TEL: 82-31-390-2411, FAX: 82-31-890-2414
e-mail: wseolha@gmail.com
†These authors contributed equally
Abstract
Leucine-rich repeat kinase 2 (LRRK2) mutations are the most common genetic cause of Parkinson's disease (PD). LRRK2 contains a functional kinase domain and G2019S, the most prevalent LRRK2 pathogenic mutation, increases its kinase activity. LRRK2 regulates mitochondria morphology and autophagy in neurons. LPS treatment increases LRRK2 protein level and mitochondrial fission in microglia, and down-regulation of LRRK2 expression or inhibition of its kinase activity attenuates microglia activation. Here, we evaluated the direct role of LRRK2 G2019S in mitochondrial dynamics in microglia. Initial observation of microglia in G2019S transgenic mice revealed a decrease in mitochondrial area and shortage of microglial processes compared with their littermates. Next, we elucidated the molecular mechanisms of these phenotypes. Treatment of BV2 cells and primary microglia with LPS enhanced mitochondrial fission and increased Drp1, a mitochondrial fission marker, as previously reported. Importantly, both phenotypes were rescued by treatment with GSK2578215A, a LRRK2 kinase inhibitor. Finally, the protein levels of CD68, an active microglia marker, Drp1 and TNF-α were significantly higher in brain lysates of G2019S transgenic mice compared with the levels in their littermates. Taken together, our data suggest that LRRK2 could promote microglial mitochondrial alteration via Drp1 in a kinase-dependent manner, resulting in stimulation of pro-inflammatory responses. This mechanism in microglia might be a potential target to develop PD therapy since neuroinflammation by active microglia is a major symptom of PD.
Graphical Abstract
Keywords: Parkinson’s disease, LRRK2, microglia, neuroinflammation, mitochondrial fission, Drp1
INTRODUCTION
Leucine rich-repeat kinase 2 (LRRK2) is a causal gene of Parkinson's disease (PD, [1,2]), which is the second most common neurodegenerative disease. LRRK2 contains functional kinase and GTPase domains. G2019S, the most prevalent LRRK2 familial pathogenic mutant, is known to exhibit increased kinase activity [3]. Most investigations into the pathogenic mechanism of LRRK2 have been focused on neuronal functions because PD is neurodegenerative disease. The studies revealed that LRRK2 regulates vesicle trafficking, autophagy, oxidative stress, neuronal toxicity and mitochondrial dynamics [4,5,6,7,8,9,10,11,12]. However, neuroinflammation with activated microglia has also been implicated in the pathogenesis of neurodegenerative disorders, such as PD, Alzheimer's disease, Huntington's disease, amyotrophic lateral sclerosis and fronto-temporal dementia [13]. LRRK2 expression in microglia is lower than in dopaminergic neurons [14]. However, protein levels of total LRRK2 and active phospho-LRRK2 were significantly increased by LPS treatment activating microglia
Mitochondria dysfunction is widely accepted as one of the pathogenic mechanisms of PD [20]. In fact, other genetic causes of PD such as mutations in Parkin and PINK1 are known to impair mitochondria [21,22]. Other studies also suggested that LRRK2 regulated mitochondrial dynamics and affected mitochondria damages in neuronal cells [12,23,24,25]. Drp1, also known as mitochondrial fission protein Dlp1, is a component of the LRRK2 interactome and is responsible for LRRK2-mediated mitochondria fission in neurons [12]. However, no study examined the effect of LRRK2 on mitochondrial morphology in microglia. Interestingly, LPS stimulations of microglia also increased mitochondrial fission, Drp1 level, production of pro-inflammatory cytokines and ROS as well as kinase activity of LRRK2 [15,16,26,27,28]. In addition, phagocytic activity of macrophages is regulated by Drp1-mediated mitochondrial fission [29]. These results suggest that LRRK2 facilitated neuroinflammatory response in microglia by regulating mitochondrial dynamics.
MATERIALS AND METHODS
The following antibodies were used: LRRK2 (N241A/34, NeuroMabs, 75-253), pS935-LRRK2 (UDD210(12), Abcam, 133450), Drp1 (C5, Santa Cruz, 271583), Tom20 (FL-145, Santa Cruz, sc-11415), CD68 (3F103, Santa Cruz, sc70761), TNFα (52B83, Santa Cruz, sc-52746), Iba-1 (Wako, 019-19741), GFAP (Sigma-Aldrich, G3839), β-actin (Santa Cruz, sc-47778), α-tubulin (DM1A, Sigma, T9026), horseradish peroxidase-conjugated goat anti-rabbit or anti-mouse IgG (Jackson ImmunoResearch. 111-035-003 or 115-035-003) and goat anti-rabbit or anti-mouse IgG H&L (Alexa Fluor® 594, Invitrogen, ab150080 or Alexa Fluor® 488, Invitrogen, ab150113).
BV2 murine microglia cells (4×105) were seeded in 35 mm dishes. Next day, the cells were treated with GSK2578215A (hereinafter GSK, 1 µM, Torcris Bioscience) for 30 min followed by LPS (1 µg/ml) treatment for 6 h. Then, the cells was fixed with 4% paraformaldehyde (PFA, Wako) for immunofluorescence analysis or harvested for Western blot analysis. Culture media derived from these BV2 cells and rat primary microglia administrated with the same treatment as BV2 cells, and brain lysates from G2019S transgenic mouse or littermate were used to perform mouse TNFα ELISA (Biolegend) or rat TNFα DuoSet ELISA (R&D systems) according to the sample species and the manufacturer's instruction.
G2019S transgenic (TG) mice [strain B6; C3-Tg (PDGFB-LRRK2*G2019S) 340D jmo/J, stock number 016575, The Jackson Laboratory, [30]] were housed in a specific pathogen-free facility at Dankook University Animal Facility on a 12:12-h light/dark cycle. Animal experiments were approved by the Institutional Animal Care and Use Committee of Dankook University (DKU-16-035). Animals were provided free access to food and water. TG mice and their normal control littermates were sacrificed by cervical dislocation. Brains were lysed with 1% Triton X-100 and protease inhibitor cocktail (Calbiochem) in phosphate buffered saline (PBS). Lysates were homogenized 10 times using a 17-gauge needle, cooled on ice for 30 min, and centrifuged at 4,000 ×g for 10 min at 4℃. Each supernatant was collected and analyzed by Western blot.
Rat primary microglial cells were isolated from neonatal rats in the p1 age as previously described [31].
G2019S TG and non-TG mice were anesthetized with pentobarbital and transcardially perfused with PBS and 4% PFA. Striatum was then dissected from each brain and subjected to EM processing as described previously [32]. Tissues were fixed in 2.5% glutaraldehyde in 0.1 M phosphate buffer (pH 7.4) for 2 h at 4℃ and post-fixed with 1% osmium tetroxide on ice for 2 h. Tissues were then embedded in Epon 812 after dehydration in an ethanol and propylene oxide series. Polymerization was conducted using pure resin at 70℃ for two days. Ultrathin sections (70 nm in thickness) were obtained with an ultramicrotome (UltraCut-UCT, Leica, Austria) and collected on 100-mesh copper grids. After staining with 2% uranyl acetate and lead citrate, the sections were examined by transmission electron microcopy (TEM) (Technai G2 Spirit TWIN, FEI, USA) at 120 kV. To quantify mitochondrial number and size, electron micrographs were obtained at a magnification of 8,000× and analyzed blindly using Image J software.
BV2 cells were treated as indicated, fixed and permeablized with 0.1 % Triton X-100 in DPBS for 5 min at RT. After 1 h of blocking with 3 % BSA and 1 % goat serum in DPBS, cells were incubated with Tom20 antibodies in blocking solution for 8 h at 4℃ followed by incubation with Alexa Fluoro 594 secondary antibodies in blocking solution at RT for 2 h with shaking. Hoechst (Thermo Scientific) staining was accomplished during the final DPBS washing step. Cells on inverted cover slip were mounted using Prolong-Gold (Invitrogen). The experiments were repeated in triplicate. Each set of experiments was observed under a Zeiss LS55 confocal microscope and more than five images were randomly obtained in Airyscan mode. In total, 10~12 cells were selected and their mitochondria (n=16~242/cell, total 412~1094) were analyzed. To determine the mitochondria area and elongation, mitochondrial morphology was measured using the Mito-Morphology Macro of Image J software as previously described [33,34]. The RFP channel of cells stained with Tom20 was extracted and converted to 8-bit grayscale, and then inverted. Individual mitochondria were highlighted on the images and analyzed for changes in mitochondrial area and circularity. The mitochondrial circularity was inversed to calculate mitochondrial elongation [34].
LRRK2 TG and non-TG mice were anesthetized as described above. Their brains were post-fixed in 4% PFA and coronally sliced into 200 µm sections using a vibratome (VT1000A, Leica, Austria). Sections were washed with PBS and treated with 3% hydrogen peroxide to block endogenous peroxidase activity. Sections were then incubated overnight with anti-Iba1 (1:500, Waco) followed by treatment with biotinylated anti-rabbit antibody (1:200, Vector Laboratories). Anti-Iba1 antibody was detected by avidin-biotin-peroxidase complex (Vector Laboratories) followed by incubation with nickel-enhanced 3, 3′diaminobenzidine (DAB, Sigma). To determine the morphological characteristics of Iba1-labeled microglia, images were obtained at 40× objective magnification using an inverted light microscope (CRX41, Olympus, Japan). The process number and length were analyzed manually using Image J software by an unbiased operator.
Harvested sample were loaded on to 11% SDS-PAGE gel or 4~15% gradient pre-cast gel (Bio-Rad) followed by transfer onto nitrocellulose membranes. The remaining steps and densitometric analysis were similar to the methods described previously [35].
Data derived from Western blots, ELISA, measured mitochondrial fission, and microglial processes were analyzed and imaged with Prism 6.0 (GraphPad). Data are expressed as means±standard error of the mean (SEM). Statistical analyses of data are described in each figure legend.
RESULTS
To investigate whether LRRK2 regulates mitochondrial fission in microglia, we compared the ultrastructure of mitochondria in the microglia of striatum in G2019S TG and control non-TG littermate mice. We confirmed that the mitochondria number was significantly increased in TG, although the total mitochondria area did not change significantly (Fig. 1A~1C). To analyze fission rates in microglia, we compared the average mitochondria areas between TG and control littermates. The average mitochondria area of TG microglia was significantly decreased compared with the non-TG control (Fig. 1D). Previous studies demonstrated shortening and decreased microglial processes in active microglia [36,37]. We observed similar phenotypes (decreased numbers and shortened length of microglial processes) of TG compared with their non-TG littermates (Fig. 2). These results support the notion that G2019S LRRK2 mutation induced microglial activation and mitochondrial fission in microglia
Previous studies revealed that treatment with ligands in microglial activation increased LRRK2 kinase activity in the primary microglia or murine BV2 microglia cell line [15,38]. Furthermore, increased mitochondrial fission by LPS in BV2 was demonstrated in a previous study [26]. To determine whether LRRK2 kinase activity directly affected mitochondrial fission in LPS-stimulated microglia, BV2 cells were treated with LPS alone or LPS and GSK2578215A (GSK, LRRK2 kinase inhibitor), and their mitochondrial morphologies were examined using an immunofluorescence assay. Image analysis revealed that LPS treatment significantly decreased mitochondrial elongation, suggesting reduced mitochondrial length [34], whereas co-treatment of LPS and GSK significantly diminished the difference (Fig. 3A, B). In addition, LPS treatment reduced mitochondrial area, and co-treatment abrogated the reduction, although without any statistical significance (Fig. 3A, C). The shortening of mitochondria in the LPS-treated BV2 cells is similar to the phenotype observed in neuronal soma in the G2019S TG [30]. These results suggest that LRRK2 regulated mitochondrial morphology in microglia as well as in neuron [30].
Previous studies reported that LRRK2 regulated Drp1 protein level in neurons [12], which prompted us to evaluate Drp1 level in microglia. Both BV2 (Fig. 4A~4F) and rat primary microglia cells (Fig. 4G~4L) were treated with LPS alone or LPS and GSK, followed by analysis of Drp1 and Tom20 protein levels. LPS treatment significantly increased the Drp1 level compared with the sample containing vehicle alone or with co-treatment with LPS and GSK whereas the level of Tom20, a mitochondria marker, was relatively similar among samples in both cell types (Fig. 4). The ratio of Drp1 level to Tom20 (Drp1/Tom20) was increased by LPS treatment compared with other samples and, as expected, the difference was more apparent in primary microglia than BV2 cells (Fig. 4E, K). Absence of significant changes in Tom20 levels suggested that the total mass of mitochondria was not significantly altered by either LPS or LPS/GSK treatment (Fig. 4D, J). However, the increased fission rate induced by LPS was reversed by co-treatment with GSK. The western blot with phospho-S935 LRRK2 antibody confirmed inhibition of LRRK2 kinase by GSK as reported previously (Fig. 4B, H, [38]). We also tested whether LPS mediated cytokine secretion to confirm activation of BV2 cells and primary microglia by LPS. LPS-mediated TNF-α secretion was significantly increased and the increase was almost restored by GSK pretreatment in both cell types (Fig. 4F, L). These data demonstrate that mitochondrial fission up-regulated by LPS stimulation was attenuated by the inhibitor of LRRK2 kinase.
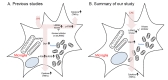
We consolidated the evidence underlying G2019S-mediated neuroinflammation and mitochondrial fission in microglia
DISCUSSION
Neurodegenerative diseases are often associated with mitochondrial dysfunction and microglial activation. In fact, PD patients show highly activated microglia in substantia nigra and enhanced pro-inflammatory cytokine expression [40,41].
Numerous studies have shown that mitochondrial dysfunction is critical in PD pathogenesis [20,22,42]. Several pathogenic mutations of PD causal genes such as Parkin, PINK-1, DJ-1, and LRRK2 are known to dysregulate mitochondrial function [22]. Treatment of BV2 with rotenone, which is a PD-causing toxin, activated microglia and generated cytokines and ROS [43] although the phenotypes were not observed in primary microglia [44]. LPS treatment also increased LRRK2 kinase activity in the brains of mice expressing FLAG-LRRK2, and treatment with LRRK2 kinase inhibitor or shLRRK2 ameliorated TNFα release [15]. Therefore, we tested whether LRRK2 regulated microglial mitochondrial function
Drp1 is a critical mediator of neuroinflammatory response [26]. LPS treatment of microglia increased mitochondrial fission, Drp1 protein level and ROS production in BV2 and mouse primary microglia [26,27]. Moreover, down-regulation of Drp1 by shDrp1 reduced mitochondrial fragmentation, cytokine production and ROS production in BV2 [26].
Our study connects LRRK2 activation and Drp1 activation, two discrete intermediate steps in LPS-mediated neuroinflammation, and suggests a signal transduction pathway from LPS to neuroinflammation via LRRK2-meiated Drp1 activation in microglia. An earlier study reported that LRRK2 phosphorylates Drp1 at T595 and the expression of T595A Drp1 corrects G2019S-mediated mitochondrial fission in neurons [45] whereas another report suggested LRRK2-mediated phosphorylation of Drp1 at S616 [46]. In addition, inhibition of Drp1 reported to protect neurons in dopaminergic system in an MPTP-mediated PD model [47], providing additional evidence for toxic mechanism of LRRK2 G2019S. Because functions of phosphorylated Drp1 are various depending on cell types and phosphorylation sites [48], it is necessary to investigate the effect of LRRK2-mediated phosphorylation of Drp1 on mitochondrial fission in microglia in a future study.
The G2019S TG mouse we used has been originally reported to express G2019S in a neuron-specific manner because the gene was under the CMV E-PDGF-β promoter [30,49]. The original report to study expression of luciferase under the same PDGF-β promoter showed that at least 15 % of cells expressing luciferase are not neurons [49], leaving a possibility for direct expression of G2019S in microglia of the TG mice. Moreover, neuronal LRRK2 expression increases secretion of α-synuclein [50] which is able to activate microglia in a paracrine manner [51]. Previous studies reported that damaged neurons activated microglia
We have previously reported that LRRK2 phosphorylates p53 at T304 and T377 residues and the expression of phosphomimic p53 in BV2 cells induces TNFα expression and its release, resulting in neuronal death [19]. The combined evidence derived from our study suggests that G2019S LRRK2 contributed to the progression of PD by altering the physical condition of mitochondria in microglia during the neuroinflammation. In addition, we suggest Drp1 as a missing link between LPS-induced LRRK2 activation and mitochondrial dynamics, both of which have been reported to induce microglial activation [15,16,26]. Furthermore, our data which was summarized in Fig. 6 suggest that LRRK2 kinase inhibitor attenuates pro-inflammatory response by rescuing of mitochondrial dysregulation in microglia. Therefore, Drp1 regulation by LRRK2 kinase in microglia may be one of the pathogenic mechanisms in PD mediated by LRRK2 G2019S mutation, indicating LRRK2 kinase as a promising PD therapeutic target.
Figures





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
References
- Paisán-Ruíz C, Jain S, Evans EW, Gilks WP, Simón J, van der Brug M, López de Munain A, Aparicio S, Gil AM, Khan N, Johnson J, Martinez JR, Nicholl D, Carrera IM, Pena AS, de Silva R, Lees A, Martí-Massó JF, Pérez-Tur J, Wood NW, Singleton AB. Cloning of the gene containing mutations that cause PARK8-linked Parkinson's disease. Neuron 2004;44:595-600.

- Zimprich A, Biskup S, Leitner P, Lichtner P, Farrer M, Lincoln S, Kachergus J, Hulihan M, Uitti RJ, Calne DB, Stoessl AJ, Pfeiffer RF, Patenge N, Carbajal IC, Vieregge P, Asmus F, Müller-Myhsok B, Dickson DW, Meitinger T, Strom TM, Wszolek ZK, Gasser T. Mutations in LRRK2 cause autosomal-dominant parkinsonism with pleomorphic pathology. Neuron 2004;44:601-607.
- West AB, Moore DJ, Biskup S, Bugayenko A, Smith WW, Ross CA, Dawson VL, Dawson TM. Parkinson's disease-associated mutations in leucine-rich repeat kinase 2 augment kinase activity. Proc Natl Acad Sci U S A 2005;102:16842-16847.
- Yun HJ, Park J, Ho DH, Kim H, Kim CH, Oh H, Ga I, Seo H, Chang S, Son I, Seol W. LRRK2 phosphorylates Snapin and inhibits interaction of Snapin with SNAP-25. Exp Mol Med 2013;45:e36.
- Shin N, Jeong H, Kwon J, Heo HY, Kwon JJ, Yun HJ, Kim CH, Han BS, Tong Y, Shen J, Hatano T, Hattori N, Kim KS, Chang S, Seol W. LRRK2 regulates synaptic vesicle endocytosis. Exp Cell Res 2008;314:2055-2065.
- Gómez-Suaga P, Fdez E, Blanca Ramírez M, Hilfiker S. A link between autophagy and the pathophysiology of LRRK2 in Parkinson's disease. Parkinsons Dis 2012;2012:324521.
- Manzoni C, Lewis PA. LRRK2 and autophagy. Adv Neurobiol 2017;14:89-105.
- Nguyen HN, Byers B, Cord B, Shcheglovitov A, Byrne J, Gujar P, Kee K, Schüle B, Dolmetsch RE, Langston W, Palmer TD, Pera RR. LRRK2 mutant iPSC-derived DA neurons demonstrate increased susceptibility to oxidative stress. Cell Stem Cell 2011;8:267-280.
- Heo HY, Park JM, Kim CH, Han BS, Kim KS, Seol W. LRRK2 enhances oxidative stress-induced neurotoxicity via its kinase activity. Exp Cell Res 2010;316:649-656.
- Smith WW, Pei Z, Jiang H, Dawson VL, Dawson TM, Ross CA. Kinase activity of mutant LRRK2 mediates neuronal toxicity. Nat Neurosci 2006;9:1231-1233.
- West AB, Moore DJ, Choi C, Andrabi SA, Li X, Dikeman D, Biskup S, Zhang Z, Lim KL, Dawson VL, Dawson TM. Parkinson's disease-associated mutations in LRRK2 link enhanced GTP-binding and kinase activities to neuronal toxicity. Hum Mol Genet 2007;16:223-232.
- Wang X, Yan MH, Fujioka H, Liu J, Wilson-Delfosse A, Chen SG, Perry G, Casadesus G, Zhu X. LRRK2 regulates mitochondrial dynamics and function through direct interaction with DLP1. Hum Mol Genet 2012;21:1931-1944.
- Amor S, Peferoen LA, Vogel DY, Breur M, van der Valk P, Baker D, van Noort JM. Inflammation in neurodegenerative diseases--an update. Immunology 2014;142:151-166.
- Miklossy J, Arai T, Guo JP, Klegeris A, Yu S, McGeer EG, Mc-Geer PL. LRRK2 expression in normal and pathologic human brain and in human cell lines. J Neuropathol Exp Neurol 2006;65:953-963.
- Moehle MS, Webber PJ, Tse T, Sukar N, Standaert DG, DeSilva TM, Cowell RM, West AB. LRRK2 inhibition attenuates microglial inflammatory responses. J Neurosci 2012;32:1602-1611.
- Kim B, Yang MS, Choi D, Kim JH, Kim HS, Seol W, Choi S, Jou I, Kim EY, Joe EH. Impaired inflammatory responses in murine Lrrk2-knockdown brain microglia. PLoS One 2012;7:e34693.
- Puccini JM, Marker DF, Fitzgerald T, Barbieri J, Kim CS, Miller-Rhodes P, Lu SM, Dewhurst S, Gelbard HA. Leucine-rich repeat kinase 2 modulates neuroinflammation and neurotoxicity in models of human immunodeficiency virus 1-associated neurocognitive disorders. J Neurosci 2015;35:5271-5283.
- Dzamko N, Rowe DB, Halliday GM. Increased peripheral inflammation in asymptomatic leucine-rich repeat kinase 2 mutation carriers. Mov Disord 2016;31:889-897.
- Ho DH, Seol W, Eun JH, Son IH. Phosphorylation of p53 by LRRK2 induces microglial tumor necrosis factor α-mediated neurotoxicity. Biochem Biophys Res Commun 2017;482:1088-1094.
- Winklhofer KF, Haass C. Mitochondrial dysfunction in Parkinson's disease. Biochim Biophys Acta 2010;1802:29-44.
- Geisler S, Holmström KM, Treis A, Skujat D, Weber SS, Fiesel FC, Kahle PJ, Springer W. The PINK1/Parkin-mediated mitophagy is compromised by PD-associated mutations. Autophagy 2010;6:871-878.
- Ryan BJ, Hoek S, Fon EA, Wade-Martins R. Mitochondrial dysfunction and mitophagy in Parkinson's: from familial to sporadic disease. Trends Biochem Sci 2015;40:200-210.
- Yang S, Xia C, Li S, Du L, Zhang L, Hu Y. Mitochondrial dysfunction driven by the LRRK2-mediated pathway is associated with loss of Purkinje cells and motor coordination deficits in diabetic rat model. Cell Death Dis 2014;5:e1217.
- Howlett EH, Jensen N, Belmonte F, Zafar F, Hu X, Kluss J, Schüle B, Kaufman BA, Greenamyre JT, Sanders LH. LRRK2 G2019S-induced mitochondrial DNA damage is LRRK2 kinase dependent and inhibition restores mtDNA integrity in Parkinson's disease. Hum Mol Genet 2017;26:4340-4351.
- Niu J, Yu M, Wang C, Xu Z. Leucine-rich repeat kinase 2 disturbs mitochondrial dynamics via Dynamin-like protein. J Neurochem 2012;122:650-658.
- Park J, Choi H, Min JS, Park SJ, Kim JH, Park HJ, Kim B, Chae JI, Yim M, Lee DS. Mitochondrial dynamics modulate the expression of pro-inflammatory mediators in microglial cells. J Neurochem 2013;127:221-232.
- Katoh M, Wu B, Nguyen HB, Thai TQ, Yamasaki R, Lu H, Rietsch AM, Zorlu MM, Shinozaki Y, Saitoh Y, Saitoh S, Sakoh T, Ikenaka K, Koizumi S, Ransohoff RM, Ohno N. Polymorphic regulation of mitochondrial fission and fusion modifies phenotypes of microglia in neuroinflammation. Sci Rep 2017;7:4942.
- Chae U, Min JS, Lee H, Song KS, Lee HS, Lee HJ, Lee SR, Lee DS. Chrysophanol suppresses pro-inflammatory response in microglia via regulation of Drp1-dependent mitochondrial fission. Immunopharmacol Immunotoxicol 2017;39:268-275.
- Wang Y, Subramanian M, Yurdagul A, Barbosa-Lorenzi VC, Cai B, de Juan-Sanz J, Ryan TA, Nomura M, Maxfield FR, Tabas I. Mitochondrial fission promotes the continued clearance of apoptotic cells by macrophages. Cell 2017;171:331-345.e22.
- Ramonet D, Daher JP, Lin BM, Stafa K, Kim J, Banerjee R, Westerlund M, Pletnikova O, Glauser L, Yang L, Liu Y, Swing DA, Beal MF, Troncoso JC, McCaffery JM, Jenkins NA, Copeland NG, Galter D, Thomas B, Lee MK, Dawson TM, Dawson VL, Moore DJ. Dopaminergic neuronal loss, reduced neurite complexity and autophagic abnormalities in transgenic mice expressing G2019S mutant LRRK2. PLoS One 2011;6:e18568.
- Lee HJ, Suk JE, Bae EJ, Lee SJ. Clearance and deposition of extracellular alpha-synuclein aggregates in microglia. Biochem Biophys Res Commun 2008;372:423-428.
- Choi KJ, Kim MJ, Je AR, Jun S, Lee C, Lee E, Jo M, Huh YH, Kweon HS. Three-dimensional analysis of abnormal ultrastructural alteration in mitochondria of hippocampus of APP/PSEN1 transgenic mouse. J Biosci 2014;39:97-105.
- Xie W, Chung KK. Alpha-synuclein impairs normal dynamics of mitochondria in cell and animal models of Parkinson's disease. J Neurochem 2012;122:404-414.
- Dagda RK, Cherra SJ, Kulich SM, Tandon A, Park D, Chu CT. Loss of PINK1 function promotes mitophagy through effects on oxidative stress and mitochondrial fission. J Biol Chem 2009;284:13843-13855.
- Ho DH, Kim H, Kim J, Sim H, Ahn H, Kim J, Seo H, Chung KC, Park BJ, Son I, Seol W. Leucine-rich repeat kinase 2 (LRRK2) phosphorylates p53 and induces p21WAF1/CIP1 expression. Mol Brain 2015;8:54.
- Hefendehl JK, Neher JJ, Sühs RB, Kohsaka S, Skodras A, Jucker M. Homeostatic and injury-induced microglia behavior in the aging brain. Aging Cell 2014;13:60-69.
- Kozlowski C, Weimer RM. An automated method to quantify microglia morphology and application to monitor activation state longitudinally
in vivo . PLoS One 2012;7:e31814.
- Marker DF, Puccini JM, Mockus TE, Barbieri J, Lu SM, Gelbard HA. LRRK2 kinase inhibition prevents pathological microglial phagocytosis in response to HIV-1 Tat protein. J Neuroinflammation 2012;9:261.
- Walker DG, Lue LF. Immune phenotypes of microglia in human neurodegenerative disease: challenges to detecting microglial polarization in human brains. Alzheimers Res Ther 2015;7:56.
- McGeer PL, Itagaki S, Boyes BE, McGeer EG. Reactive microglia are positive for HLA-DR in the substantia nigra of Parkinson's and Alzheimer's disease brains. Neurology 1988;38:1285-1291.
- Sawada M, Imamura K, Nagatsu T. Role of cytokines in inflammatory process in Parkinson's disease. J Neural Transm Suppl 2006;70:373-381.
- Abou-Sleiman PM, Muqit MM, Wood NW. Expanding insights of mitochondrial dysfunction in Parkinson's disease. Nat Rev Neurosci 2006;7:207-219.
- Gao F, Chen D, Hu Q, Wang G. Rotenone directly induces BV2 cell activation via the p38 MAPK pathway. PLoS One 2013;8:e72046.
- Klintworth H, Garden G, Xia Z. Rotenone and paraquat do not directly activate microglia or induce inflammatory cytokine release. Neurosci Lett 2009;462:1-5.
- Su YC, Qi X. Inhibition of excessive mitochondrial fission reduced aberrant autophagy and neuronal damage caused by LRRK2 G2019S mutation. Hum Mol Genet 2013;22:4545-4561.
- Esteves AR, G-Fernandes M, Santos D, Januário C, Cardoso SM. The upshot of LRRK2 inhibition to Parkinson's disease paradigm. Mol Neurobiol 2015;52:1804-1820.
- Filichia E, Hoffer B, Qi X, Luo Y. Inhibition of Drp1 mitochondrial translocation provides neural protection in dopaminergic system in a Parkinson's disease model induced by MPTP. Sci Rep 2016;6:32656.
- Hu C, Huang Y, Li L. Drp1-dependent mitochondrial fission plays critical roles in physiological and pathological progresses in mammals. Int J Mol Sci 2017;18:144.
- Liu BH, Wang X, Ma YX, Wang S. CMV enhancer/human PDGF-β promoter for neuron-specific transgene expression. Gene Ther 2004;11:52-60.
- Kondo K, Obitsu S, Teshima R. α-Synuclein aggregation and transmission are enhanced by leucine-rich repeat kinase 2 in human neuroblastoma SH-SY5Y cells. Biol Pharm Bull 2011;34:1078-1083.
- Kim C, Ho DH, Suk JE, You S, Michael S, Kang J, Joong Lee S, Masliah E, Hwang D, Lee HJ, Lee SJ. Neuron-released oligomeric α-synuclein is an endogenous agonist of TLR2 for paracrine activation of microglia. Nat Commun 2013;4:1562.
- Sudo S, Tanaka J, Toku K, Desaki J, Matsuda S, Arai T, Sakanaka M, Maeda N. Neurons induce the activation of microglial cells
in vitro . Exp Neurol 1998;154:499-510.
- Witting A, Müller P, Herrmann A, Kettenmann H, Nolte C. Phagocytic clearance of apoptotic neurons by Microglia/Brain macrophages
in vitro : involvement of lectin-, integrin-, and phosphatidylserine-mediated recognition. J Neurochem 2000;75:1060-1070.