Articles

Article Tools
 View Full Text View Full Text |
 Abstract Abstract |
 Article as PDF Article as PDF |
 Print this Article Print this Article |
 Pubmed Pubmed |
 PMC PMC |
 PubReader PubReader |
 Export to Citation Export to Citation |
 Email Alerts Email Alerts |
 Open Access Open Access |
Share this article on :



Stats or Metrics
Article
Review Article
Exp Neurobiol 2018; 27(5): 321-343
Published online October 31, 2018
https://doi.org/10.5607/en.2018.27.5.321
© The Korean Society for Brain and Neural Sciences
Sex-specific Behavioral Features of Rodent Models of Autism Spectrum Disorder
Se Jin Jeon1,2, Edson Luck Gonzales2,3, Darine Froy N. Mabunga2,3, Schley T. Valencia2,3, Do Gyeong Kim2,3, Yujeong Kim2,3, Keremkleroo Jym L. Adil2,3, Dongpil Shin2,3, Donghyun Park2,3, and Chan Young Shin2,3,4*
1Center for Neuroscience, Korea Institute of Science & Technology, Seoul 02792, Korea.
2Department of Pharmacology and Advanced Translational Medicine, School of Medicine, Konkuk University, Seoul 05029, Korea.
3Department of Neuroscience, School of Medicine and Center for Neuroscience Research, Konkuk University, Seoul 05029, Korea.
4KU Open Innovation Center, Konkuk University, Seoul 05029, Korea.
Correspondence to: *To whom correspondence should be addressed.
TEL: 82-2-2030-7834, FAX: 82-2-2030-7899
e-mail: chanyshin@kku.ac.kr
Abstract
Sex is an important factor in understanding the clinical presentation, management, and developmental trajectory of children with neuropsychiatric disorders. While much is known about the clinical and neurobehavioral profiles of males with neuropsychiatric disorders, surprisingly little is known about females in this respect. Animal models may provide detailed mechanistic information about sex differences in autism spectrum disorder (ASD) in terms of manifestation, disease progression, and development of therapeutic options. This review aims to widen our understanding of the role of sex in autism spectrum disorder, by summarizing and comparing behavioral characteristics of animal models. Our current understanding of how differences emerge in boys and girls with neuropsychiatric disorders is limited: Information derived from animal studies will stimulate future research on the role of biological maturation rates, sex hormones, sex-selective protective (or aggravating) factors and psychosocial factors, which are essential to devise sex precision medicine and to improve diagnostic accuracy. Moreover, there is a strong need of novel strategies to elucidate the major mechanisms leading to sex-specific autism features, as well as novel models or methods to examine these sex differences.
Graphical Abstract
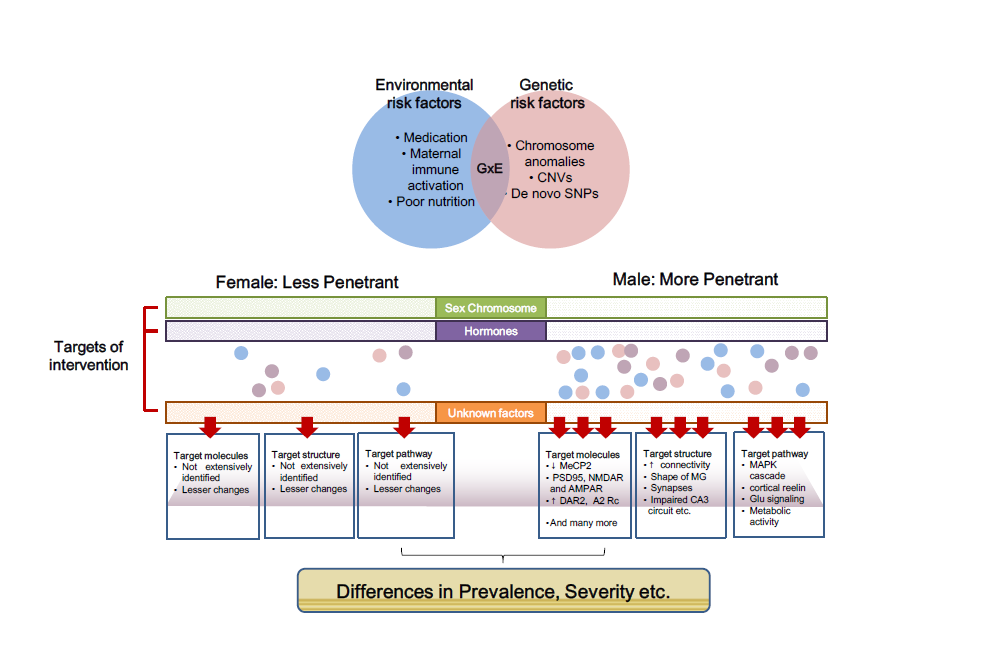
Keywords: Sex difference, Autism spectrum disorder, Animal model, Behavior
INTRODUCTION
From as early as the second day of conception, signs of sex differences in development are already apparent [1] and the intricate process that follows in embryonic development makes these differences even more complex. In addition to the outwardly apparent differences in the reproductive organs and other body parts such as hair, skin and even facial features, sex differences in brain development and function have also been observed. At the physical level, male and female brains display volumetric, structural and anatomical differences in various regions across life stages [2,3,4]. Moreover, also physiologic studies reveal instances functional sex differences in the brain, by showing that some cognitive abilities, equally performed between males and females, have sex-differential patterns and levels of brain activation. For example, males and females can solve certain math problems equally, but the patterns and levels of brain activation might be higher in females than males [3,5]. Factors such as chromosomal differences and the resulting differential expression of genes that are transcribed to express molecules such as sex hormones could shape the sexual dimorphisms described above, but no clear mechanism has been presented. Besides, there is ample evidence that factors other than sex hormones are also involved in sex differences in brain function [6,7], which makes delineating the sex difference issue in normal and pathological conditions a formidable challenge. Lovell-Badge and Robertson first described the four core genotypes (FCG) mice, characterized by the deletion of the testis-determining gene Sry from the Y chromosome (XY females) and the insertion of an Sry gene into the autosomes (XX males) [8,9]. Thus, the FCG mice produce XX and XY gonadal males and XX and XY gonadal females. The juvenile social play in FCG mice has been studied to determine sex differences and chromosomal effects in social interaction behaviors. In normal C57BL/6J mice, non-sibling, samesex pairing revealed that males have a tendency to be more social than females, as manifested by close physical contact with their partners, while, on the other hand, female mice spent more time engaged in anogenital sniffing and play solicitation than males. In FCG mice, both wild-type male and female mice (XX females and XY males) have greater social behaviors than their heterochromosomal counterparts (XY females and XX males), and the XY male group showed the highest social behavior compared to any other group. Indeed, the interaction of sex chromosomes and gonadal sex affects the social behavior in FCG mice, with the possible additional influence of sex hormones in social behaviors [10]. Other neurobehavioral features such as aggression [11], parenting [12], nociception [13], and drug abuse [14] observed in FCG mice indicate that sex differences in such behaviors is partly due to a sex chromosome effect as well.
Remarkably, some disease conditions have sexual dimorphism at least in the prevalence rate, although the underlying causes are still unexplained. In some cases of autoimmune diseases like systemic lupus erythematosus, multiple sclerosis or Sjögren's syndrome, there is a marked skewness in the prevalence toward females [15,16], while in neurodevelopmental disorders like language impairments and attention deficit hyperactivity disorder (ADHD), the prevalence is higher in males [17,18]. ASD is one of the neurodevelopmental disabilities with male preponderance. However, the higher male prevalence in ASD also remains an unsolved puzzle. Recent efforts are geared towards understanding the mechanistic underpinnings of sex differences in ASD.
According to the Center for Disease Control and Prevention (CDC), the male-to-female prevalence ratio of ASD is estimated to be about 4.5 [19]. In a recent meta-analysis of prevalence studies from 54 publications, the general male-to-female odds ratio (MFOR) was estimated to be 4.20, while stricter conditions and selection of high-quality studies only gave an MFOR of about 3.1 [20]. The discrepancy between these two values could be attributed to the apparent diagnostic bias between sexes, and specifically to a tendency not to clinically diagnose females who meet the ASD diagnosis criteria. Interestingly, females have fewer externalizing problems and restricted and repetitive behaviors than males, suggesting that the discrepancy in phenotypes between males and females with ASD may contribute to the biased prevalence [21,22]. The impression is that the female population, characterized by fewer diagnoses of ASD, may display weaker manifestations of the core and accompanying symptoms of autism, and that a higher genetic liability is required to induce full-blown autism in females [22,23]. Efforts to explain the differences have identified a potential protective mechanism in females against
Another prevailing hypothesis to explain the sex differences in autism is the “Empathizing-Systemizing (ES) Theory of Psychological Sex Differences” [25]. This theory provides evidence that males have higher systemizing scores, while females have higher empathizing scores, and that these tendencies make the male more susceptible to social impairments when exposed to environmental insults or genetic mutations during the early developmental period. Empathy involves an emotional response to another individual's behavior, whereas systemizing represents a more intellectual and logical approach to understanding the objects and the environment [26]. Thus, the greater empathizing ability of females may alleviate the social alterations, so that a higher percentage of diagnosed or undiagnosed females with ASD, especially those with average IQ scores, have better socialization and communication abilities than their male counterparts [27,28]. One underlying factor possibly explaining the male-specific features is fetal testosterone during development, which has a negative correlation with social behaviors but a positive correlation with restricted interests [29].
In line with clinical findings, the study of sex difference in ASD animal models will be an important tool to elucidate various mechanistic aspects of the differences. However, interest in this area increased only recently, since most of the preclinical studies typically used only males. Factors such as hormonal differences, especially the estrus cycle of females, are just one of the reasons encouraging the more frequent use of males in research. However, the presentation of sex differences in behavior underscores the need to include female animals in basic research studies. Similarly, sex differences in the presentation, treatment, and outcomes of mental illnesses are often overlooked. Keeping in mind the theories putatively explaining the sex difference in human ASD development, this review is concerned in how researchers have tried to address the sex difference of ASD in animal models, and whether the results support the theories or not. We look at the current progress of sex difference studies in animal models of ASD, and identify the areas where most improvement is needed in future work.
SEX DIFFERENCES IN ENVIRONMENTAL MODELS OF AUTISM
The valproic acid (VPA)-exposed animal model is one of the most widely used models of ASD because of its known clinical relevance and validity [30,31,32]. Most studies using this model have predominantly exploited the male sex, and few studies have delved into the sex differences in behaviors and brain molecular changes. Clinically, the occurrence of ASD in children exposed to VPA
The animal studies, in rat models, which took into account possible sex differences found that social impairment, increased electric-shock induced seizure susceptibility, reduced pain sensitivity and increased anxiety-like behaviors were significantly observed in the male offspring prenatally exposed to VPA but not in their female littermates [33,34,35]. On the other hand, increased repetitive/stereotyped behaviors was observed in both male and female VPA-exposed rats [35]. In the visuospatial attention and sensorimotor gating behaviors, both male and female VPA rats showed similar levels of abnormalities with minor differences in specific parameters [36]. Mice exposed to VPA also exhibited a male-prevalent social deficit, manifested by reduced sniffing of a stranger mouse in the three-chamber assay, although both sexes have similar abnormalities in the open field and elevated plus maze tasks [37]. Combined together, the experimental results so far suggest that VPA male animals have greater impairment in social interaction, while other behaviors may or may not be different, suggesting that social interaction is the most sensitive behavioral domain affected in a sex-specific manner. Therefore, a female protective or compensatory mechanism towards the development of normal social capability could be modulating the sex differences in VPA rodent models.
Interestingly, the prefrontal cortex region of VPA-exposed rat offspring revealed a male-specific reduction of methyl CpG binding protein 2 (MeCP2) expression from as early as embryonic day 14 (two days after VPA injection), which was sustained up to the second week of postnatal period [33,38]. This phenomenon could also have driven the male-biased increased levels in the expression of postsynaptic density protein 95 (PSD95), N-Methyl-D-aspartate (NMDA) receptor subunits, and α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptor subunits during the neonatal and juvenile period of development, which has important implications on the excitatory-inhibitory imbalance in the affected brain [33,38]. In mice, neuronal cell loss in the prefrontal cortex and transient increase in apoptotic-like cell death in the neocortex were observed following exposure to VPA during the embryonic period for both sex [37]. These results suggest that differences in the sophisticated mechanisms governing neural differentiation or synaptic development, but not the general massive cellular toxicity against ASD-inducing stimuli (in this case, prenatal VPA exposure) underlie the observed sex differences in behavioral characteristics.
Similarly, RNA-sequencing and proteomics analysis were performed in prenatally VPA-exposed male juvenile rats, and showed reduced expression of genes such as
Another context that could be involved in the sex differences displayed by the VPA rat model is the neuroendocrine and immune system. According to Schneider and colleagues [35], male VPA rats have reduced thymus weight, decreased proliferation response of splenocytes to mitogenic stimulation, and increased NO production by peritoneal macrophages in the basal and lipopolysaccharide (LPS) stimulated states, whereas a decreased IFN-γ/IL-10 ratio was noted in both sexes. In addition, male, but not female, VPA rats have elevated corticosterone levels versus controls under basal conditions. Indeed, some of the findings of immune dysregulation in the VPA rat model have also been observed in autistic children [40], and it has been suggested that the neuroimmune system together with masculinization factors, such as sex hormones, may make males more vulnerable to developing ASD [41]. In addition, several lines of evidence suggest that VPA can affect both the reproductive and the endocrine systems, including sex hormone levels, of men and women [42,43,44,45]. Thus, the sexual dimorphism in the endocrine and immune response of rats to VPA exposure may be an interesting area to help explain the unresolved sex bias of autism prevalence in humans. More interestingly, a recent review by McCarthy et al. [46] suggested that sexual dimorphism in brain development could be mediated by neuroinflammatory dynamics, which might be driven by divergent neuroepigenetic profiles between sexes. Future studies in these areas will provide insight into the role of immunology in various neurodevelopmental disorders like ASD and in the unresolved issue of sex differences.
Brain metabolic activities in VPA rat models have been studied recently by Cho and colleagues [34] using independent component analysis (ICA) of cross-sectional Positron Emission Tomography (PET) data to look at changes in metabolic networks. This method revealed changes caused by VPA treatment in the metabolic activity of the various brain regions examined, showing decreased activities of the olfactory bulb and thalamus, and increased activity of the left caudoputamen in both male and female rats when compared to their respective controls [34]. In addition, some areas of connectivity between brain regions were affected by VPA treatment in both sexes, including those connecting the olfactory bulb to the nucleus accumbens, the somatosensory cortex to the insular cortex, and the olfactory bulb to the orbitofrontal cortex and the cerebellum. Interestingly, sex differences in the effect of VPA treatment on brain connectivity, but not metabolic activity, were found. These include the connections between visual cortex and cerebellum, and between retrosplenial cortex, nucleus accumbens and olfactory bulb, which were less increased in VPA-exposed males than in VPA-exposed females; and connections between anterodorsal hippocampus and caudoputamen, and between medulla and insular/motor cortex, that were more increased in VPA-exposed males than females. Thus, the metabolic activity and connectivity changes caused by VPA may clarify the connection between the behavioral and cellular alterations in the pathophysiology of ASD, and reveal subtle differences in the sex bias aspect of ASD. How similar changes in metabolic activity can result in differential brain connectivity needs to be further investigated in future studies.
Taken together, the VPA animal model of ASD continues to be a useful tool to help understand yet unknown pathways in the development of ASD and the differences between male and female mechanisms, and especially in explaining the differences in behavioral outcomes such as social, emotional and cognitive function.
Zinc is an essential micronutrient with an important role in brain development processes such as neurogenesis, neuronal differentiation, and migration, as well as synaptic plasticity [47,48]. Zinc deficient mice models have been generated due to the possible association between zinc deficiency and autism [49,50], wherein zincdependent Shank scaffold proteins and the zinc-metalloprotease-BDNF axis could be strongly involved in the pathophysiology [51,52]. Animals who were zinc-deficient during the prenatal period exhibited behavioral abnormalities such as anxiety-like behaviors, impaired social-related behaviors, aggression, and altered cognition [53].
One research group addressed possible sex differences in the prenatal zinc deficiency (PZD) mice model [54]. During the neonatal period, the PZD mice showed reduced zinc levels in the brain, which however started to normalize after being cared for by foster mothers. Thus, at the time of experiment, during adulthood, the PZD mice already had a normal range of zinc levels in the brain. Behaviorally, the PZD mice have increased anxiety, reduced nest-building capability, delayed latency to call but a normal number of calls during a reciprocal social interaction test, and decreased latency to fall during the rotarod test, which were not sex-specific. Notably, both male and female PZD mice have normal sociability behaviors, normal spontaneous ability during the Y maze test and normal self-grooming behaviors. Concerning sex differences, the females show increased freezing behavior during transfer to a new environment or cage, more significant reduction of nest-building capacity, impaired preference to social novelty, reduced oral-to-oral interaction during reciprocal social interaction, and reduced marble burying behavior. On the other hand, male PZD mice seem to have longer sniffing duration in response to a social stimulus than control males. In general, according to this study, the few sex-specific effects of PZD seem to have more negative effects in female than male mice. This is surprising, considering that most of the sex difference effects in many autism models are more prevalent in males. Moreover, there are weak abnormalities or even enhancing effects of PZD on social and self-grooming behaviors, especially in males, suggesting that more studies will be needed to clearly validate this model of ASD. Although the PZD model sometimes shows contradictory social and self-grooming behaviors, unexpected in an ASD models, the causative relationship of zinc deficiency to other etiologic factors of ASD, such as Shank proteins, make it an interesting model to delve into the mechanistic examination of sex differences, and to examine whether zinc deficiency is more debilitating in the female autistic population.
Maternal infection causing immune activation during pregnancy is one of the many environmental risk factors for ASD [55,56,57,58]. Both viral and bacterial infections were associated with increased risk of ASD development in the offspring [59,60]. To strengthen the involvement of maternal immune activation (MIA) in ASD development, the measurement of an inflammatory biomarker, C-reactive protein (CRP), in the mothers of autistic and normal children found direct proportionality between CRP levels and autism risk [61]. Thus, the development of MIA model in animals eventually emerged, using immunogens such as LPS and polycytidylic acid (poly (I:C)) to mimic bacterial and viral infections, respectively [62]. The use of MIA as animal models of ASD has been widely discussed in the literature [63]. Here, we gathered the few studies that have specifically tackled the sex-difference effects of MIA models in ASD core and accompanying symptoms. The offspring of C57BL/6J mice prenatally treated with both LPS and/or poly (I:C) during the gestation period displayed behavioral impairments with treatment and sex-specific effects [64,65,66]. Foley and colleagues [67] reported the sex-differential effects of prenatal exposure to lipopolysaccharide (LPS), or pre- and postnatal exposure to propionic acid (PPA), in the acoustic startle response and prepulse inhibition in Long-Evans rats. Prenatal LPS exposure resulted in hypersensitivity to acoustic startle only in male offspring, while both pre- and postnatal exposure to PPA induced initial changes in startle response in both male and female rats. Meanwhile, a female-biased deficit in prepulse inhibition was observed in PPA-exposed offspring. Overall, these studies concluded that prenatal, postnatal and the combination of pre- and postnatal exposures to LPS or PPA can induce subtle and sex-differential effects in the sensory processing of adolescent rats.
Xuan and Hampson [64] found that the locomotor activity of MIA-exposed mice was reduced in both sex for poly (I:C) but not LPS exposure. In the three-chamber social test paradigm, female mice exposed to both immunogens showed reduced preference towards a social stimulus, while males were socially affected in poly (I:C) but not in LPS-exposed conditions [64]. Furthermore, marble burying was increased only in males exposed to both immunogens, but females were not affected, and grooming behaviors were not changed in either sex [64]. In contrast, Ruskin et al. [66] showed that poly (I:C) exposure could reduce social interaction and increase grooming behavior in male but not female mice. Yet another study by Hui and colleagues [65] revealed that prenatal poly (I:C) exposure could impair social interaction and increase marble burying in both male and female offspring, whereas increased anxiety and decreased prepulse inhibition were observed only in males. Furthermore, one study found no, or at most few, sex-specific effects of MIA exposure in autism-relevant behaviors [68]. These differences in behavioral phenotypes among studies could be explained by the variability in dosage and timing of poly (I:C) injection during pregnancy. Therefore, agreement between results and methods among researchers would be needed to better understand sex differences in the effects of MIA in autism, and to maximize the translational perspective [63]. Of particular interest is the idea that the interaction of genetic liability with environmental factors (such as MIA) could be another promising area of investigation for the study of sex difference in ASD [69]. Only a few studies have attempted to elucidate the possible sex differences in the effects of MIA at the molecular and structural level. In the study by Hui et al. [65], prenatal exposure to poly (I:C) affected structural and molecular features in the brain in a sex-dependent manner. In the hippocampus, altered morphology and distribution of the microglia were noted in male but not female mice. Moreover, alterations in the expression of pro-inflammatory genes both in the cerebral cortex, cerebellum, and hippocampus were mostly male-biased [65]. Indeed, more in-depth mechanistic studies are needed to understand the pathophysiologic implications of the differences in relevant behaviors between male and female MIA-exposed animals. It should also be noted that MIA has been introduced as a ‘neurodevelopmental disease primer’, indicating that it can have broad effects and induce other neurologic conditions as well [56].
SEX DIFFERENCE IN GENETIC MODELS OF AUTISM
The
MeCP2, generally regarded as a transcriptional repressor, binds methylated CpG dinucleotides, resulting in the tightening of the chromatin coil leading to a decrease in transcription. It also recruits histone deacetylases (HDACs) [78,79]. However, depending on the temporal and tissue context, it was also recently shown to behave as either a repressor or a translational activator, enhancing the expression of 85% of its target genes [80,81,82]. Most of MeCP2's binding sites are associated with active transcription, with only a small percentage of these sites being methylated cytosines [82].
Mutations of MeCP2 were shown to be the genetic cause of Rett syndrome [83]. However, these mutations are also associated with other phenotypically similar neurodevelopmental disorders [84,85,86], indicating the need for a correct MeCP2 dosage to ensure neuronal functional integrity [87,88,89,90]. The protein has been suggested to play a vital role in modulating changes in gene expression in response to neuronal activity [91]. For example, MeCP2 phosphorylation regulates activity-dependent BDNF expression [92,93]. Its phosphorylation at S421 is dependent on neuronal activity, and is necessary for MeCP2-modulated dendritic growth and spine maturation [92].
Expression of MeCP2 was found to be sex-, and time-dependent. During the critical period for brain sexual differentiation in rodents, approximately between embryonic day 18 (E18) and postnatal day 10 (PN10) [94,95], females expressed more MeCP2 in the amygdala and hypothalamus (at PN1). This difference in expression levels finally equalizes between the sexes by day 10 (PN10) [96]. In another study, this difference in protein expression in rat prefrontal cortex (PFC) was observed from E14 to PN7, but did not differ between sex at PN14 [97]. Interestingly, mutations in the MeCP2 gene were found in autism [91,98,99,100], albeit they are not common [101,102].
These discoveries prompted researchers to ask whether disruption of this sex-specific MeCP2 expression is responsible for the male bias observed in autism. In a study conducted by Kurian et al. [103], a transient knockdown of MeCP2 protein by siRNA in the amygdala of female rats revealed no changes in play behavior; however, knockdown in male rats resulted into a significant decline of juvenile social play behavior, which resembles femaletypical play behavior. While both male and female rats were observed to have reduced MeCP2 expression, altered juvenile social behavior was only evident in males. This male-specific sensitivity to MeCP2 disruption during neonatal period provides an intriguing link to the observed male bias associated with reduced MeCP2 expression, which may again point to the so-called female protective mechanism against disruptive stimuli for neural development and synaptic function.
Moreover, there are sex-dependent features of the transcriptome in MeCP2 knockout (KO) mice. Our transcriptome analysis comparing male and female mice model of MeCP2 KO, available from Gene Expression Omnibus (GEO) (Male datasets: GSE105045; Female datasets: GSE90736) revealed that there are 149 differentially expressed gene (DEGs) for male mice and 430 DEGs for female mice, with a base 2 logarithmic fold changes greater than 1.5 (p-value<0.04, Fig. 1A). For both datasets we used 22–24 weeks old MeCP2 KO mice. RNA sequencing was performed with Illumina HiSeq. In the GSE105045 and GSE90736 datasets we analyzed the transcriptome of cortex tissue and microglia, respectively. We explored the mouse phenotypes and Gene Ontology categories shared by common DEGs. Phenotypes enriched in both male and female KO models were mostly related to morphological categories. The Gene Ontology analysis performed with the Database for Annotation, Visualization and Integrated Discovery (DAVID), showed that DEGs shared by both male and female mice were, among biological processes, mainly related to transport processes (Fig. 1B). Phenotypes from male DEGs were enriched in those related to the increase of body weight (Fig. 1C). One study reported that male MeCP2 hypomorphic mice showed significantly increased body weight from 9 weeks of age [104]. On the other hand, most DEGs from female MeCP2 KO mice had a phenotypical link to decreased body weight (Fig. 1D), and it is known clinically that female MeCP2 patients have lower weight than normal people [105]. Gene Ontology (GO) analysis also revealed that DEGs from male MeCP2 KO mice were significantly associated with sensory perception of pain (Fig. 1C). Interestingly, it was shown that sensory pain perception can be regulated by MeCP2 [106]. In addition, Gene Ontology results indicated angiogenesis as one of the most strongly enriched biological processes in female MeCP2 KO mice (Fig. 1D). Experimental results on senescent endothelial progenitor cells (EPCs) revealed that MeCP2 might moderate angiogenesis [107]. These analytical results suggest that the transcriptomic differences are closely related to the phenotypic sex differences in an animal model of MeCP2.
Based on evidence that ASD is one of the many neurodevelopmental disorders involving excitatory/inhibitory imbalance (E/I imbalance), Meng et al. [108] investigated the effects of MECP2 on specific neuronal types. Male mice without MECP2 in their glutamatergic neurons, induced through conditional knockout, were observed to display tremors, anxiety-related behaviors, abnormal seizure-like brain activity and severe obesity. On the other hand, female Mecp2-heterozygous mice were less affected, and were shown to have normal lifespans [109]. Accordingly, only female mice demonstrated relief from ataxia when MeCP2 was restored in glutamatergic neurons [108].
A decrease in excitatory synapses was also observed in MECP2 shRNA-transfected neurons [110]. In a separate study, transgenic male mice displayed an increased number of glutamatergic synapses compared to MECP2-null mice [88]. Likewise, Kim et al. [97] traced the male preponderance of the VPA animal model of autism to the influence of MeCP2 in postsynaptic development. Interestingly, they hypothesized that MeCP2 downregulation was the effect of the proteasomal degradation properties of VPA, which again may be dependent on its HDACi activity, based on previous studies reporting VPA to have induced degradation of some proteins via the proteasomal pathway [111,112]. On the other hand, an increase in NR2A mRNA level was observed in both cerebellum and hypothalamus in MECP2-deficient male mice [80,113]. Similarly, increased expressions of excitatory synaptic protein marker PSD-95 and neural stem cell marker Tuj-1 were also observed in cultured rat neural progenitor cells transfected with siRNA against MeCP2 [97]. However, the absence of MECP2 also altered the basal inhibitory rhythms of the hippocampal CA3 circuit, resulting in increased susceptibility to hyperexcitability [114]. These contradicting results imply MECP2's capacity to broadly regulate excitatory neuronal development and function, which recommends specific investigations of the role of this important epigenetic regulator in a neuronal context-dependent manner to gain a finer understanding of its sex-specific functions related to ASD.
MeCP2 is also linked to the regulation of vasopressin (AVP) expression in the hypothalamus [115]. AVP, in itself, is associated with a number of social behaviors [116,117,118] and AVP in the amygdala is vital in governing these social behaviors [119]. In the mammalian brain, higher levels of AVP are found in the male than in the female amygdala [120], implying it as a potential target of MeCP2 in modulating social behavior in males [121]. This was shown in male rats with transient reduction of MeCP2 levels in their amygdala during their first three days of postnatal life. A decrease of AVP expression was observed in the first two weeks of life, which persisted to adulthood. This was not demonstrated in their female counterparts [122]. Male brains appear to be more sensitive to changes in molecules associated with epigenetic regulation during early brain development. These findings call for further investigations of the epigenetic mechanisms regulating sexual differentiation of the brain, which would clarify why there is a male-bias in the susceptibility to autism, while females appear to be more resilient to the disorder.
Euchromatin histone methyltransferase 1 (EHMT1 or G9a-like protein, GLP), together with its paralog, EHMT2 (G9a), forms a chromatin remodeling complex that catalyzes the dimethylation of histone H3 at lysine 9 (H3K9me2), a post-translational modification associated with repression of gene transcription [123]. H3K9me2 is an epigenetic mark that is dynamically regulated in the hippocampus and nucleus accumbens during contextual fear memory formation, addiction, and stress [124]. Additionally, Ehmt1 is highly expressed in the embryonic brain, and in restricted areas of the adult brain, providing a plausible mechanism through which EHMT1 can affect neurodevelopmental processes and, as a result, higher cognitive functions.
Kleefstra syndrome (KS) is a neurodevelopmental disorder caused by the haploinsufficiency of the EHMT1 gene. KS is characterized by intellectual disability, general developmental delay, childhood hypotonia, craniofacial abnormalities and autistic-like behavioral problems. In addition, mutations in EHMT1 have also been associated with ASD [125] and schizophrenia [126]. Many of the human clinical features of KS are recapitulated in mice carrying constitutive haploinsufficiency of Ehmt1 (Ehmt1+/−), making it a highly valid mammalian model for the human disease [127,128].
Both male and female Ehmt1+/− mice showed increased anxiety, decreased sniffing and approaching time to novel objects or mice, and normal locomotor activity that was not sex-specific [129]. The sex differences observed in these mice include decreased juvenile playing time in adolescent males, and prolonged and delayed preference for social novelty in adult males, but no preference for social novelty in adult females. Male mice showed no preference for stranger mice, but such preference was recovered for a period of time. On the contrary, the females showed no preference for the stranger during all the test sessions, implying that female mice might be more severely affected in their social preference compared to their male counterparts. On the other hand, both male and female mice seem to display more sniffing time in presence of social stimulus than in the empty compartment, indicating higher sociability compared to the wild-type mice [129]. Another core symptom of autism, stereotyped/repetitive behavior, was not clearly confirmed in Ehmt1+/− mice since they showed no differences in spontaneous T maze alternation compared to the wildtype group [129].
Since EHMT1 is required for H3K9me2-mediated gene repression, impaired H3K9me2 by EHMT1 deficiency is suspected to be the main cause of the autism-like behaviors as well as KS [130]. Expression of BDNF and Rab3b is known to be affected by H3K9me-2 mediated gene repression, which mediates homeostatic excitatory and inhibitory synaptic plasticity both in vitro and in vivo, respectively [131,132,133]. Thus, altered dosage of those targets might be one of the etiological factors of autism and KS. Notably, the special importance of H3K9me2 and EHMT1 expression in male germ cell development and function was reported [134,135]. In male mice, EHMT1 is expressed from intermediate spermatogonia until the pre-leptotene stage, and H3K9me2 persists during the synapsis stage. On the other hand, in females, EHMT1 is expressed until the leptotene, and the H3K9me2 methyl marks are maintained during the diplotene. Post pachytene male germ cells complete the meiosis and differentiate into sperm rapidly, while female meiosis stops at the diplotene stage and lasts until ovulation. Thus, male-specific rapid turnover of H3K9me2 at the pachytene stage might be a driving force for the sex-specific features of development. Another intriguing possibility is that EHMT1 is recruited by the transcriptional repressor element-1 (RE1) silencing transcription factor (REST), and assembled with other corepressors such as HDAC1, HDAC2, and the Rett-syndrome-related protein MECP2 onto the promoters of target genes modulating epigenetic remodeling and gene repression [136,137]. MECP2 is also known to be implicated in sex differences in neurodevelopment (more details are discussed in the MeCP2 section). The possibility that EHMT1 and MeCP2 play a cooperative role in the regulation of neural development implies a crucial role of epigenetic regulation in ASD and in sex differences.
Methylenetetrahydrofolate reductase (Mthfr) plays a role in processing amino acids, the building blocks of proteins, and in the production of the vitamin folate (also called vitamin B9) [138]. Specifically, this enzyme converts a molecule called 5,10-methylenetetrahydrofolate to a molecule called 5-methyltetrahydrofolate [138]. This reaction is required for the multistep process that converts the amino acid homocysteine to another amino acid, methionine. Defects in Mthfr gene regulation and abnormal homocysteine-folate metabolism have been reported to increase the risk of birth defects such as neural tube defects, oral clefts, and Down syndrome [139]. Furthermore, an increased risk has been reported for neuropsychiatric and neurodegenerative diseases such as depression, obsessive-compulsive disorder, Alzheimer's disease, Parkinson's disease, schizophrenia, and autism [140,141].
In mice, because of lethality in the homozygotes, behavioral features were confirmed using Mthfr+/− mice. They showed hyperactivity, anxiety, cognitive deficits and low sociability in both sex [142,143]. However, the anxiety level in the open field task and social preference deficit was much higher in females compared to males, which might be related to the dysregulated excitatory and inhibitory synaptic protein levels. Interestingly, Mthfr deficiency-mediated enhancement of cortical reelin and glutamatergic signaling in female mice might be a major factor mediating social and anxiety-related problems [143]. These effects would be affected by sex hormones that influence synaptogenesis and synaptic plasticity, such as estradiol [144]. Estradiol recruits PSD95, neuroligin 1, and the NMDA receptor subunit GluN1 to the dendrite spine, resulting in increased excitatory synapses and altering the excitatory/inhibitory neurotransmitter balance in primary neurons [144]. Notably, this sexual hormone-induced synaptogenesis works in female but not male mice [145], but the underlying mechanism is unclear. This may also explain the sex-dependent enhancement of excitatory synapses in Mthfr+/− females. Another plausible mechanism is GABA signaling, which is directly regulated by estradiol [146]. Blumkin et al. [143] showed the sex-dependent GAD activation and potentiation of the GABAergic system in Mthfr+/− females, suggesting a deficit in basal inhibitory function. Overall, sex-specific Mthfr deficiency is critical for the excitatory and inhibitory synaptic plasticity balance that affects cognition and social behavior. Even though the sex differences in neural development may be context-specifically regulated, depending on the etiological factor involved, at least it is obvious that recurrent pathological findings in many neurodevelopmental psychiatric disorders such as E/I imbalance are preserved in Mthfr+/− mice. However, the results from the Mthfr+/− male and females mice clearly indicate that sex selective neural development and autistic behaviors are not invariably biased in one direction, in which females are always protected, but may be governed by the complex interplay of etiological factors, each of which may have differential roles in males and females.
The BTBR T+tf/J mouse model is an inbred strain, commonly used to study ASD, which, according to previous studies, exhibits behavioral phenotypes with face validity to all three diagnostic symptom categories of autism: lower reciprocal social interactions as juveniles and adults, lower social approach behaviors, reduced social transmission of food preference, unusual patterns of ultrasonic vocalizations, and high repetitive self-grooming as compared to the C57BL/6J strain (B6) [147,148]. Only a few studies have examined possible sex differences in this strain. One such study was undertaken to examine the effect of Poly IC, a substance which induces maternal immune activation, in C57/BL6 and BTBR mice. Male BTBR mice showed ASD-like symptoms such as social approach, ultrasonic vocalization, marble burying and self-grooming behaviors significantly more than female mice [149].
In another sociability study, male B6 and female BTBR mice displayed a preference for sex- and strain-matched conspecific stimulus while their sex counterpart did not [150]. Although there was no significant interaction between sex and strain in the social proximity test, a significant main effect of sex indicated that female mice showed higher levels of frontal contact (nose tip-to-nose tip) and lower levels of posterior investigation (nose-to-anogenital) in comparison to male mice, altogether suggesting different motivations for sociability in males and females. Systemic administration of the anxiolytic diazepam decreased the frequency of two behaviors associated with anxiety and defensiveness, upright and jump escape, as well as crawl-under behavior [150]. These findings may be interpreted as similar to the gaze aversion response that is commonly reported in autistic individuals, contributing to the view that BTBR mice constitute an excellent model for the analysis of autistic-like behaviors. These results suggest that the BTBR model can be used to study social aspects of autism, including sex-specific behaviors.
Glutathione (GSH) is the most abundant low molecular weight thiol [151]. It is a tripeptide formed by the amino acids L-glutamate, L-cysteine and L-glycine, capable of inhibiting damage to essential cell components caused by reactive oxygen species, such as heavy metals, peroxides, liquid peroxides, and free radicals [152,153]. Atypical expression of genes of the oxidative stress pathways and an increase in oxidative stress have been reported in ASD [154,155]. Phenotypic differences of genes associated with the metabolism of glutathione, e.g. GSTP and GSTM1 (glutathione-S-transferaseM1), have been correlated with autistic spectrum disorder [156].
GSTM1 is found in humans and in mice [157]. The deletion or removal of GSTM1 may cause an organism to become more susceptible to environmental insults. Therefore, when combined with an environmental insult, alternative GSTM1 genotypes may result in alterations of neurological development and behaviors that mimic those seen in individuals with autism [158].
Yochum et al. [159] used this model to determine if genetically altered mice are more sensitive to toxicant exposure early in life. They used mice with GSTM1 deletion and wild-type controls exposed to valproic acid on PN14. As expected, both GTSM1 knockout male and female mice demonstrated behavioral phenotypes reminiscent of that of autism. Considering that female GTSM1 knockout mice also showed autistic phenotypes, deletion of GTSM1 may also leave females vulnerable to VPA-induced neurotoxicity. Interestingly, female GTSM1 wild-type mice treated with VPA had a decreased number of apoptotic cells from TUNEL staining at P14, suggesting that a neuroprotective effect is exerted in females against VPA damage. Of note, differences in the expression of GTSM1 between sex, with increased expression in females, are observed in both liver and colon in human and mouse [160,161]. A significantly higher incidence of the GSTM1-null genotype was found in female multiple sclerosis patients compared with male patients, and it has been suggested that GSTM1's role may possibly be sex-dependent [162]. These studies point out that GSTM1 may exert a role in neural cell survival in female mice during early development, making them less susceptible to environmental insults compared to male mice. Overall, the neurobehavior of GSTM1 knockout mice shows sex- and stage-specific difference and further mechanistic studies are needed.
A clinical study on individuals diagnosed with ASD revealed deletions and duplications in chromosomal region 16p11.2 [163]. These alterations, which occur with frequency similar to that of the duplication of the Prader-Willi/Angelman region, are a common cause of autism and predispose the individual to neurodevelopmental disorders including ADHD [164,165]. To further uncover the possible mechanisms and pathophysiology of the disorder and explain the sex-difference in striatal dysfunction, a mouse model of 16p11.2 hemideletion was produced [166]. According to Ted Abel's group, del/+ males, but not females, displayed deficits in reward-directed learning and maintaining motivation to work for rewards, accompanied by increased striatal ERK1 phosphorylation [166]. Since ERK1 is involved in functional repression of striatum, while ERK2 has the opposite effect [167], ERK1 up-regulation probably induced the operant learning impairment which is, on the other hand, not manifested by female del/+ animals. Since ERK1 is located within the 16p11.2 region, the mRNA of ERK1 was reduced by half in del/+ animals [166]. However, the striatal ERK1 in del/+ males, but not in females, is hyperphosphorylated under basal conditions, and this effect is significantly exacerbated by reward, implying that ERK1 phosphorylation is linked to the goal-directed learning deficit in the striatum, in a sex dependent manner. The underlying mechanism which causes sex differences in the ERK pathway in the striatum remains unclear, but there are several candidates including protein phosphatase-kinase intracellular signaling, and the Y-chromosome specific USP9Y protein ubiquitination/degradation factor [168].
Another significant finding in their study is the overexpression of the mRNA of dopamine receptor 2 and adenosine receptor 2a in the striatum in male animals with the deletion. These proteins are regarded as markers of medium spiny neurons signaling via the indirect pathway, which is linked to behavioral inhibition. Hence, in this case, the obvious influence of sex in the genetic lesion was connected with neurodevelopmental disorders, involving intracellular signaling mechanisms in the brain to explain the male-specific susceptibility and female-specific protective ability [166].
Additional information about sex-biased behavioral deficits was sought by examining whether 16p11.2 del/+ animals would exhibit sex-specific sleep and activity alterations, two co-morbid symptoms present in some neurodevelopmental disorders, including ASD and ADHD [169]. The data revealed home-cage hyperactivity in both sexes, but sex-biased alterations in sleep tests. 16p11.2 del/+ male, but not female, mice were observed to spend significantly more time awake and significantly less time in nonrapid eye movement (NREM) sleep during the 24 h period than their wild-type littermates [169].
Beside the male-specific behavioral deficits exhibited by the mouse model of 16p11.2 hemideletion (del/+), another study investigated sex-specific neuroanatomical endophenotypes, due to the commonly accepted notion that brain structural changes are a relevant factor in the pathogenesis of neurodevelopmental disorders [170]. Twenty-seven genes are deleted in del/+ animals, and their expression patterns were analyzed using the Allen Mouse Brain Atlas, showing that they spatially overlapped the brain regions affected by structural changes [171]. These results confirmed an apparent elevation of fractional anisotropy in the medial fiber tracts proximal to the striatum in male del/+ mice alone, supporting the previous study of sex-specific differences [166]. Interestingly, the genes overexpressed in regions of the male del/+ endophenotypic changes are associated with the regulation of neurite outgrowth and the activity of MAPK pathway, two credible mechanisms for the pathogenesis of neurodevelopmental disorders, suggesting the interconnection between neural activity and neuroanatomical endophenotypic changes in the sex-biased regulation of ASD-like behaviors [171].
Chromodomain-helicase-DNA binding protein 8 (CHD8) is one of the chromatin remodeling proteins that can regulate various autism risk genes [172,173,174]. It has been reported that heterozygous mutations of CHD8 in males show the behavioral features of ASD [175,176]; however, the involvement of sex dimorphism has not been investigated in detail except in one recent study [177]. Jung et al. [177] showed male-biased behavioral abnormalities in social communication in pups, mother attachment behaviors in juvenile, and increased self-grooming in adults, but these phenotypes did not appear in females. The authors demonstrates that neural activity is suppressed in the female mice under both basal and stress conditions, but the male CHD8 mutants exhibited normal baseline activity and excessive neural activity under the stress condition.
Moreover, transcriptomic analysis showed many GO terms underlying the sex-dependent features in CHD8 mutants such as extracellular matrix (ECM), that regulates the development, function, and plasticity of synapses [177,178]. The ECM-related genes in the females showed upregulation, but downregulation in the males, which results in the differential neural activity as shown by the c-Fos activity. Collectively, male-biased ASD behavioral properties in CHD8 mutant mice are linked to the sexual DEGs in the transcriptome, and to differential neuronal activities, making these mice a useful tool, especially for the sex-specific behavioral changes regulated by gene expression.
Hu et al. [179,180] identified a number of genes with dysregulated expression through gene expression and methylation profiling of lymphoblastoid cell lines (LCL) from monozygotic twins and pairs of siblings discordant for ASD diagnosis. RAR-related orphan receptor alpha (Rora), a nuclear hormone receptor which serves as a transcriptional regulator, was among the many to be found hypermethylated and consequently downregulated [179,180]. Decreased Rora expression was also observed in the prefrontal cortex and cerebellum of these patients [181]. Rora has been associated with circadian rhythm regulation [182], which is disrupted in patients with ASD [183]. In addition, Rora directly regulates
Rora plays a role in the translational regulation of CYP19A1 [191], which codes for aromatase, an enzyme responsible for converting testosterone to estradiol [192]. Aromatase is considered crucial in modulating male and female sex hormone levels in various tissues, including the brain [193]. Reciprocally, estradiol was found to upregulate RORA expression, while the androgen dihydrotestosterone (DHT) downregulates it [193].
Correlation studies have revealed that Rora and a number of its transcriptional targets may display sex-dependent expression in certain brain regions of both humans and mice [194]. Investigation of the correlation coefficients between Rora expression and its transcriptional target genes (
Interestingly, Sarachana and colleagues [193] reported decrements of Rora in the brain tissue of both male and female subjects with ASD and suggested that while
While the above-mentioned studies seem to implicate the disruption of Rora expression and its transcriptional targets in the sex differences in autism, there were no studies so far directly demonstrating that dysregulated Rora expression could result in the development of autism-related behavioral phenotypes. Prenatal hormonal disruption and the resulting Rora dysregulation should also be considered in a context of gene-environment interplay [197], where biological sex acts as a regulator. Epidemiologically-based endocrine studies listed increased prenatal steroidogenic activity as an early ‘environmental’ risk for the later diagnosis of autism in males [198]. Etiological investigation for autism should also focus on such context, considering the male-specific environmental risk factors that were identified, and the proposed correlation between genes and environment.
As we described in the previous sections, some of the genes related to autism are found in the sex chromosomes and some may also indirectly result in sex-specific consequences. We also identified environmental factors whose effects are seemingly skewed towards the male sex. While the contributing genes vary in different patients and many gene variants at different loci are necessary for the disorder to develop, the concentration of most of the suggested environmental risk factors is not enough to induce the disease [199]. These facts suggest a possible interaction or perhaps synergism between genetic and environmental factors in increasing the risk for autism and its reported male preponderance.
Schaafsma et al. [69] put the “triple hit” (gene×environment×male sex) hypothesis to the test by inducing maternal immune activation in a contactin-associated protein-like 2 (Cntnap2) mouse model, and investigated the autism-like effects in male and female animals, thus reproducing the three “hits” of ASD-related etiological factors. These hits demonstrated cumulative effects on ultrasonic vocalization and resulted in deficits in social behavior. The synergistic effects of these hits also significantly altered the expression of the corticotropin-releasing hormone receptor-1 (Crh1) in the left hippocampus, while simultaneously altering histone H3 N-terminal lysine 4 trimethylation (H3K4me3). These findings suggest that both gene and environmental factors may cooperate with male-biased biological mechanism to determine the final outcome of ASD phenotypes. This hypothesis, while increasingly gaining recognition, still needs further exploration, in particular of the quantitative contribution of each factor to the manifestation of ASD-like traits.
CONCLUSIONS
Since the origin of research on sex differences, it has been hypothesized that sex differences in behavior are at least in part caused by hormonal influences on the brain [200]. Of course, not all sex differences can be attributed to sex hormones, but genetic, epigenetic, and chromosomal effects also play an important role [201]. At the moment, it is very important to note that sex per se (and probably the sex hormone levels as well) and the interaction with the genetic predisposition and environmental stimuli govern the final outcomes of neural development and synaptic function, which eventually affect sex differences in behavior.
In addition, studying sex-dependent innate behaviors such as sociability, cognition or empathy is useful to understand the underlying mechanisms of sex- or circumstance-dependent neurobiological behaviors in mice. Using a model of pre-gestational stress, a study explored the effects of perinatal exposure to fluoxetine, a selective serotonin reuptake inhibitor, on social play behavior and the hypothalamic pituitary adrenal (HPA) axis. Perinatal administration of fluoxetine resulted in diminished negative effect of maternal stress on sibling play behavior, but increased social aggressive play with a novel conspecific in both sexes and decreased grooming of a novel conspecific in males. Furthermore, it increased serum corticosteroid binding globulin levels, hippocampal serotonin levels, and pre-synaptic density probed using synaptophysin in the dentate gyrus. Pre-gestational maternal stress, in itself, produced a decline in hippocampal neurogenesis rates and synaptophysin density in the dentate gyrus of pre-adolescent males, but not females [202]. This study demonstrated how pregestational maternal stress, perinatal selective serotonin reuptake inhibitors (SSRIs), and sex exert their effects on the developing social behavior and the related hippocampal plasticity of the preadolescent offspring.
Known as major modulators of the excitatory/inhibitory balance in the brain, glutamate and GABA signaling systems play a vital role in ASD pathophysiology, and these neurotransmission systems are not free from sex biases. Both glutamate and GABA regulation are influenced by sex hormones. In fact, studies reported the neuroprotective effects of female sex hormones, prolactin (PRL) and estrogen, against hippocampal neurodegeneration caused by glutamate excitotoxicity [203,204,205]. Furthermore, progesterone acts on non-NMDA receptors (AMPA and kainite) and suppresses glutamatergic excitatory activity (reviewed in [206]). Al-Suwailem et al. [205] demonstrated a significantly lower level of glutamate in the brains of female Wistar albino rats than their male counterparts and suggested that the male bias observed in autism may be explained by the reduced susceptibility of females to glutamate excitotoxicity. Upon the onset of testicular activity in males, estradiol, converted from testosterone by aromatase, initializes a cascade of differentiating effects on the neuronal substrate, giving rise to the masculine brain [207]. Estradiol has the capacity to both boost and prolong the duration of the initial developmental excitatory effects of GABA [207]. On the other hand, progesterone enhances GABAergic inhibitory transmission through its interaction with GABAA receptors in the mature brain (reviewed in [206]).
Indeed, animal studies also reflect the sexually dimorphic regulation of glutamatergic and GABAergic signaling. In our study [208], we observed aberrations in the kinetic profile of NMDAR, AMPAR, and mGluR5 pathways in the prefrontal cortex of VPA-exposed male rats. These observations were coupled with a reduction of MeCP2 expressions in both the prefrontal cortex of the male offspring and neuronal progenitor cells isolated by sex. We observed the same male bias in the TERT-tg mice, which shows autistic-like phenotypes, more pronounced increase in the expression of VGluT1, a presynaptic marker, and increases in postsynaptic markers of the NMDA and AMPA receptor subtypes [209].
As we described in the previous sections (Table 1), the strong male preponderance in ASD together with a greater genetic load in female ASD led to a female protective effect (FPE) hypothesis [210]. Similarly, sex chromosomes and hormones have been suggested as the underlying mechanisms. However, the basic mechanisms of sex-specific phenotypes from many animal models leave plenty of room for further investigation. For example, experiments using sex chromosome aneuploidy models, or genetic and pharmacological gonadal steroid perturbation models, in combination with ASD models may provide better insight into the role of sex chromosomes and hormones in sex-biased ASD manifestation. Obviously, the unsolved questions in the field of sex skewness of ASD may enormously benefit from experimental results, which might need large number of biological replicates, but not much from merely speculative assumptions about the differences in the structural, functional, and behavioral domains of neural correlates of sex. Overall, combining the knowledge from cellular, molecular, and animal studies on how sex, together with other parameters, influences neural development and neurological functions such as altered neurotransmitter systems, we can expand our understanding and examine the underlying hypotheses, including FPE, about sex-specific neurodevelopmental and psychiatric behaviors and improve diagnosis and intervention through increased precision.
Figures

{kind=link}
Tables
{kind=link}
| ASD Model | Male phenotype | Female phenotype | References |
|---|---|---|---|
| VPA | Reduced sociability and social recognition | Repetitive behavior | Kim et al., 2013 |
| Increased seizure susceptibility | Cho et al., 2017 | ||
| Reduced pain sensitivity | Schneider et al., 2008 | ||
| Increased anxiety | |||
| Repetitive behavior | |||
| PZD | Increased sociability | Reduced nest-building | Grabrucker et al., 2016 |
| Increased anxiety | Impaired social novelty | ||
| Decreased social interaction | |||
| Repetitive behavior | |||
| Increased anxiety | |||
| MIA | Abnormal startle response | Abnormal startle response | Xuan et al., 2014 |
| Reduced social preference | Social preference deficit | Hui et al., 2018 | |
| Increased repetitive behavior | Ruskin et al., 2017 | ||
| Hyperactivity | Foley et al., 2015 | ||
| MeCP2 | Declined juvenile social play | Abnormal motor function | Kurian et al., 2008 |
| Anxiety-like behavior | Breathing problem | Meng et al., 2016 | |
| Abnormal seizure-like tremor | Patterson et al., 2016 | ||
| Motor abnormality | |||
| Ehmt1 | Anxiety-like behavior | Anxiety-like behavior | Balemans et al., 2010 |
| Reduced sociability and preference | Decreased sociability and novelty | ||
| Decreased juvenile social play | No social novelty preference | ||
| Mthfr | Anxiety | Anxiety-like behavior | Levav-Rabkin et al., 2011 |
| Hyperactivity | Hyperactivity | Blumkin et al., 2011 | |
| Cognition problem | Cognitive deficit | ||
| Reduced social preference | Decreased social preference | ||
| BTBR | USV problem | Mild USV problem | Schwartzer et al., 2013 |
| Repetitive behavior | Mild repetitive behavior | ||
| Reduced sociability | Mild reduced sociability | ||
| Gstm1 | Repetitive behavior | Mild repetitive behavior | Yochum et al., 2010 |
| Reduced sociability | Mild reduced sociability | Singhal et al., 1992 | |
| 16p11.2 hemideletion | Reward-directed learning deficit | Hyperactivity | Grissom et al., 2018 |
| Hyperactivity | Angelakos et al., 2017 | ||
| Sleep deprivation | |||
| CHD8 | Social communication problem | No specific behavior features | Jung et al., 2018 |
| Hypoactivity | Platt et al., 2017 | ||
| Repetitive behavior | |||
| RORA | Repetitive behavior | Not determined in mouse model | Goodall et al., 1987 |
| Reduced exploration | Lalonde et al., 2003 |
VPA, valproic acid; PZD, prenatal zinc deficiency; MIA, maternal immune activation; MeCP2, Methyl CpG binding protein 2; Ehmt1, Euchromatin histone methyltransferase 1; Mthfr, Methylenetetrahydrofolate reductase; BTBR, BTBR T+tf/J; Gstm1, glutathione-S-transferaseM1; CHD8, Chromodomain-helicase-DNA-binding protein 8; RORA, RAR-related orphan receptor alpha.
References
- Ray PF, Conaghan J, Winston RM, Handyside AH. Increased number of cells and metabolic activity in male human preimplantation embryos following
in vitro fertilization. J Reprod Fertil 1995;104:165-171.
- Giedd JN, Vaituzis AC, Hamburger SD, Lange N, Rajapakse JC, Kaysen D, Vauss YC, Rapoport JL. Quantitative MRI of the temporal lobe, amygdala, and hippocampus in normal human development: ages 4–18 years. J Comp Neurol 1996;366:223-230.
- Lenroot RK, Giedd JN. Sex differences in the adolescent brain. Brain Cogn 2010;72:46-55.
- De Bellis MD, Keshavan MS, Beers SR, Hall J, Frustaci K, Masalehdan A, Noll J, Boring AM. Sex differences in brain maturation during childhood and adolescence. Cereb Cortex 2001;11:552-557.
- Bell EC, Willson MC, Wilman AH, Dave S, Silverstone PH. Males and females differ in brain activation during cognitive tasks. Neuroimage 2006;30:529-538.
- Ritchie SJ, Cox SR, Shen X, Lombardo MV, Reus LM, Alloza C, Harris MA, Alderson HL, Hunter S, Neilson E, Liewald DC, Auyeung B, Whalley HC, Lawrie SM, Gale CR, Bastin ME, McIntosh AM, Deary IJ. Sex differences in the adult human brain: evidence from 5216 UK biobank participants. Cereb Cortex 2018;28:2959-2975.
- Floris DL, Lai MC, Nath T, Milham MP, Di Martino A. Network-specific sex differentiation of intrinsic brain function in males with autism. Mol Autism 2018;9:17.
- Lovell-Badge R, Robertson E. XY female mice resulting from a heritable mutation in the primary testis-determining gene, Tdy. Development 1990;109:635-646.
- Mahadevaiah SK, Odorisio T, Elliott DJ, Rattigan A, Szot M, Laval SH, Washburn LL, McCarrey JR, Cattanach BM, Lovell-Badge R, Burgoyne PS. Mouse homologues of the human AZF candidate gene RBM are expressed in spermatogonia and spermatids, and map to a Y chromosome deletion interval associated with a high incidence of sperm abnormalities. Hum Mol Genet 1998;7:715-727.
- Cox KH, Rissman EF. Sex differences in juvenile mouse social behavior are influenced by sex chromosomes and social context. Genes Brain Behav 2011;10:465-472.
- Maxson SC, Didier-Erickson A, Ogawa S. The Y chromosome, social signals, and offense in mice. Behav Neural Biol 1989;52:251-259.
- Gatewood JD, Wills A, Shetty S, Xu J, Arnold AP, Burgoyne PS, Rissman EF. Sex chromosome complement and gonadal sex influence aggressive and parental behaviors in mice. J Neurosci 2006;26:2335-2342.
- Gioiosa L, Chen X, Watkins R, Klanfer N, Bryant CD, Evans CJ, Arnold AP. Sex chromosome complement affects nociception in tests of acute and chronic exposure to morphine in mice. Horm Behav 2008;53:124-130.
- Quinn JJ, Hitchcott PK, Umeda EA, Arnold AP, Taylor JR. Sex chromosome complement regulates habit formation. Nat Neurosci 2007;10:1398-1400.
- Whitacre CC. Sex differences in autoimmune disease. Nat Immunol 2001;2:777-780.
- Patel R, Shahane A. The epidemiology of Sjögren's syndrome. Clin Epidemiol 2014;6:247-255.
- Ramtekkar UP, Reiersen AM, Todorov AA, Todd RD. Sex and age differences in attention-deficit/hyperactivity disorder symptoms and diagnoses: implications for DSM-V and ICD-11. J Am Acad Child Adolesc Psychiatry 2010;49:Array-Array.
- Black LI, Vahratian A, Hoffman HJ. Communication disorders and use of intervention services among children aged 3–17 years: United States, 2012. NCHS Data Brief 2015;:1-8.
- Christensen DL, Baio J, Van Naarden Braun K, Bilder D, Charles J, Constantino JN, Daniels J, Durkin MS, Fitzgerald RT, Kurzius-Spencer M, Lee LC, Pettygrove S, Robinson C, Schulz E, Wells C, Wingate MS, Zahorodny W, Yeargin-Allsopp M, Centers for Disease Control and Prevention (CDC). Prevalence and characteristics of autism spectrum disorder among children aged 8 years--Autism and Developmental Disabilities Monitoring Network, 11 Sites, United States, 2012. MMWR Surveill Summ 2016;65:1-23.
- Loomes R, Hull L, Mandy WP. What is the male-to-female ratio in autism spectrum disorder? A systematic review and meta-analysis. J Am Acad Child Adolesc Psychiatry 2017;56:466-474.
- Werling DM, Geschwind DH. Sex differences in autism spectrum disorders. Curr Opin Neurol 2013;26:146-153.
- Szatmari P, Liu XQ, Goldberg J, Zwaigenbaum L, Paterson AD, Woodbury-Smith M, Georgiades S, Duku E, Thompson A. Sex differences in repetitive stereotyped behaviors in autism: implications for genetic liability. Am J Med Genet B Neuropsychiatr Genet 2012;159B:5-12.
- Jacquemont S, Coe BP, Hersch M, Duyzend MH, Krumm N, Bergmann S, Beckmann JS, Rosenfeld JA, Eichler EE. A higher mutational burden in females supports a “female protective model” in neurodevelopmental disorders. Am J Hum Genet 2014;94:415-425.
- Robinson EB, Lichtenstein P, Anckarsäter H, Happé F, Ronald A. Examining and interpreting the female protective effect against autistic behavior. Proc Natl Acad Sci U S A 2013;110:5258-5262.
- Baron-Cohen S, Knickmeyer RC, Belmonte MK. Sex differences in the brain: implications for explaining autism. Science 2005;310:819-823.
- McClure I. The essential difference: men, women and the extreme male brain. BMJ 2003;327:57.
- Halladay AK, Bishop S, Constantino JN, Daniels AM, Koenig K, Palmer K, Messinger D, Pelphrey K, Sanders SJ, Singer AT, Taylor JL, Szatmari P. Sex and gender differences in autism spectrum disorder: summarizing evidence gaps and identifying emerging areas of priority. Mol Autism 2015;6:36.
- Head AM, McGillivray JA, Stokes MA. Gender differences in emotionality and sociability in children with autism spectrum disorders. Mol Autism 2014;5:19.
- Knickmeyer R, Baron-Cohen S, Raggatt P, Taylor K. Foetal testosterone, social relationships, and restricted interests in children. J Child Psychol Psychiatry 2005;46:198-210.
- Mabunga DF, Gonzales EL, Kim JW, Kim KC, Shin CY. Exploring the validity of valproic acid animal model of autism. Exp Neurobiol 2015;24:285-300.
- Christensen J, Grønborg TK, Sørensen MJ, Schendel D, Parner ET, Pedersen LH, Vestergaard M. Prenatal valproate exposure and risk of autism spectrum disorders and childhood autism. JAMA 2013;309:1696-1703.
- Rasalam AD, Hailey H, Williams JH, Moore SJ, Turnpenny PD, Lloyd DJ, Dean JC. Characteristics of fetal anticonvulsant syndrome associated autistic disorder. Dev Med Child Neurol 2005;47:551-555.
- Kim KC, Kim P, Go HS, Choi CS, Park JH, Kim HJ, Jeon SJ, Dela Pena IC, Han SH, Cheong JH, Ryu JH, Shin CY. Male-specific alteration in excitatory post-synaptic development and social interaction in pre-natal valproic acid exposure model of autism spectrum disorder. J Neurochem 2013;124:832-843.
- Cho H, Kim CH, Knight EQ, Oh HW, Park B, Kim DG, Park HJ. Changes in brain metabolic connectivity underlie autistic-like social deficits in a rat model of autism spectrum disorder. Sci Rep 2017;7:13213.
- Schneider T, Roman A, Basta-Kaim A, Kubera M, Budziszewska B, Schneider K, Przewłocki R. Gender-specific behavioral and immunological alterations in an animal model of autism induced by prenatal exposure to valproic acid. Psychoneuroendocrinology 2008;33:728-740.
- Anshu K, Nair AK, Kumaresan UD, Kutty BM, Srinath S, Laxmi TR. Altered attentional processing in male and female rats in a prenatal valproic acid exposure model of autism spectrum disorder. Autism Res 2017;10:1929-1944.
- Kataoka S, Takuma K, Hara Y, Maeda Y, Ago Y, Matsuda T. Autism-like behaviours with transient histone hyperacetylation in mice treated prenatally with valproic acid. Int J Neuropsychopharmacol 2013;16:91-103.
- Cho H, Kim CH, Knight EQ, Oh HW, Park B, Kim DG, Park HJ. Changes in brain metabolic connectivity underlie autistic-like social deficits in a rat model of autism spectrum disorder. Sci Rep 2017;7:13213.
- Barrett CE, Hennessey TM, Gordon KM, Ryan SJ, McNair ML, Ressler KJ, Rainnie DG. Developmental disruption of amygdala transcriptome and socioemotional behavior in rats exposed to valproic acid prenatally. Mol Autism 2017;8:42.
- Ashwood P, Wills S, Van de Water J. The immune response in autism: a new frontier for autism research. J Leukoc Biol 2006;80:1-15.
- McCarthy MM, Wright CL. Convergence of sex differences and the neuroimmune system in autism spectrum disorder. Biol Psychiatry 2017;81:402-410.
- Verrotti A, Mencaroni E, Cofini M, Castagnino M, Leo A, Russo E, Belcastro V. Valproic acid metabolism and its consequences on sexual functions. Curr Drug Metab 2016;17:573-581.
- Taubøll E, Gregoraszczuk EL, Tworzydø A, Wójtowicz AK, Ropstad E. Comparison of reproductive effects of levetiracetam and valproate studied in prepubertal porcine ovarian follicular cells. Epilepsia 2006;47:1580-1583.
- Taubøll E, Gregoraszczuk EL, Kołodziej A, Kajta M, Ropstad E. Valproate inhibits the conversion of testosterone to estradiol and acts as an apoptotic agent in growing porcine ovarian follicular cells. Epilepsia 2003;44:1014-1021.
- Aldemir E, Akdeniz F. Effects of valproate on male reproductive functions. Turk Psikiyatr Derg 2009;20:376-384.
- McCarthy MM, Nugent BM, Lenz KM. Neuroimmunology and neuroepigenetics in the establishment of sex differences in the brain. Nat Rev Neurosci 2017;18:471-484.
- Levenson CW, Morris D. Zinc and neurogenesis: making new neurons from development to adulthood. Adv Nutr 2011;2:96-100.
- Takeda A, Nakamura M, Fujii H, Tamano H. Synaptic Zn(2+) homeostasis and its significance. Metallomics 2013;5:417-423.
- Grabrucker AM. Environmental factors in autism. Front Psychiatry 2013;3:118.
- Yasuda H, Yoshida K, Yasuda Y, Tsutsui T. Infantile zinc deficiency: association with autism spectrum disorders. Sci Rep 2011;1:129.
- Koh JY, Lim JS, Byun HR, Yoo MH. Abnormalities in the zinc-metalloprotease-BDNF axis may contribute to megalencephaly and cortical hyperconnectivity in young autism spectrum disorder patients. Mol Brain 2014;7:64.
- Grabrucker S, Jannetti L, Eckert M, Gaub S, Chhabra R, Pfaender S, Mangus K, Reddy PP, Rankovic V, Schmeisser MJ, Kreutz MR, Ehret G, Boeckers TM, Grabrucker AM. Zinc deficiency dysregulates the synaptic ProSAP/Shank scaffold and might contribute to autism spectrum disorders. Brain 2014;137:137-152.
- Hagmeyer S, Haderspeck JC, Grabrucker AM. Behavioral impairments in animal models for zinc deficiency. Front Behav Neurosci 2015;8:443.
- Grabrucker S, Boeckers TM, Grabrucker AM. Gender dependent evaluation of autism like behavior in mice exposed to prenatal zinc deficiency. Front Behav Neurosci 2016;10:37.
- Hisle-Gorman E, Susi A, Stokes T, Gorman G, Erdie-Lalena C, Nylund CM. Prenatal, perinatal, and neonatal risk factors of autism spectrum disorder. Pediatr Res 2018;84:190-198.
- Estes ML, McAllister AK. Maternal immune activation: implications for neuropsychiatric disorders. Science 2016;353:772-777.
- Jiang HY, Xu LL, Shao L, Xia RM, Yu ZH, Ling ZX, Yang F, Deng M, Ruan B. Maternal infection during pregnancy and risk of autism spectrum disorders: a systematic review and meta-analysis. Brain Behav Immun 2016;58:165-172.
- Lee BK, Magnusson C, Gardner RM, Blomström Å, Newschaffer CJ, Burstyn I, Karlsson H, Dalman C. Maternal hospitalization with infection during pregnancy and risk of autism spectrum disorders. Brain Behav Immun 2015;44:100-105.
- Atladóttir HÓ, Thorsen P, Østergaard L, Schendel DE, Lemcke S, Abdallah M, Parner ET. Maternal infection requiring hospitalization during pregnancy and autism spectrum disorders. J Autism Dev Disord 2010;40:1423-1430.
- Zerbo O, Qian Y, Yoshida C, Grether JK, Van de Water J, Croen LA. Maternal infection during pregnancy and autism spectrum disorders. J Autism Dev Disord 2015;45:4015-4025.
- Brown AS, Sourander A, Hinkka-Yli-Salomäki S, McKeague IW, Sundvall J, Surcel HM. Elevated maternal C-reactive protein and autism in a national birth cohort. Mol Psychiatry 2014;19:259-264.
- Lombardo MV, Moon HM, Su J, Palmer TD, Courchesne E, Pramparo T. Maternal immune activation dysregulation of the fetal brain transcriptome and relevance to the pathophysiology of autism spectrum disorder. Mol Psychiatry 2018;23:1001-1013.
- Careaga M, Murai T, Bauman MD. Maternal immune activation and autism spectrum disorder: from rodents to nonhuman and human primates. Biol Psychiatry 2017;81:391-401.
- Xuan IC, Hampson DR. Gender-dependent effects of maternal immune activation on the behavior of mouse offspring. PLoS One 2014;9:e104433.
- Hui CW, St-Pierre A, El Hajj H, Remy Y, Hébert SS, Luheshi GN, Srivastava LK, Tremblay MÈ. Prenatal immune challenge in mice leads to partly sex-dependent behavioral, microglial, and molecular abnormalities associated with schizophrenia. Front Mol Neurosci 2018;11:13.
- Ruskin DN, Murphy MI, Slade SL, Masino SA. Ketogenic diet improves behaviors in a maternal immune activation model of autism spectrum disorder. PLoS One 2017;12:e0171643.
- Foley KA, MacFabe DF, Kavaliers M, Ossenkopp KP. Sexually dimorphic effects of prenatal exposure to lipopolysaccharide, and prenatal and postnatal exposure to propionic acid, on acoustic startle response and prepulse inhibition in adolescent rats: relevance to autism spectrum disorders. Behav Brain Res 2015;278:244-256.
- Schwartzer JJ, Careaga M, Onore CE, Rushakoff JA, Berman RF, Ashwood P. Maternal immune activation and strain specific interactions in the development of autism-like behaviors in mice. Transl Psychiatry 2013;3:e240.
- Schaafsma SM, Gagnidze K, Reyes A, Norstedt N, Månsson K, Francis K, Pfaff DW. Sex-specific gene-environment interactions underlying ASD-like behaviors. Proc Natl Acad Sci U S A 2017;114:1383-1388.
- Belichenko NP, Belichenko PV, Li HH, Mobley WC, Francke U. Comparative study of brain morphology in Mecp2 mutant mouse models of Rett syndrome. J Comp Neurol 2008;508:184-195.
- Zoghbi HY. MeCP2 dysfunction in humans and mice. J Child Neurol 2005;20:736-740.
- Kankirawatana P, Leonard H, Ellaway C, Scurlock J, Mansour A, Makris CM, Dure LS, Friez M, Lane J, Kiraly-Borri C, Fabian V, Davis M, Jackson J, Christodoulou J, Kaufmann WE, Ravine D, Percy AK. Early progressive encephalopathy in boys and MECP2 mutations. Neurology 2006;67:164-166.
- Carter JC, Lanham DC, Pham D, Bibat G, Naidu S, Kaufmann WE. Selective cerebral volume reduction in Rett syndrome: a multiple-approach MR imaging study. AJNR Am J Neuroradiol 2008;29:436-441.
- Coy JF, Sedlacek Z, Bächner D, Delius H, Poustka A. A complex pattern of evolutionary conservation and alternative polyadenylation within the long 3″-untranslated region of the methyl-CpG-binding protein 2 gene (MeCP2) suggests a regulatory role in gene expression. Hum Mol Genet 1999;8:1253-1262.
- Aber KM, Nori P, MacDonald SM, Bibat G, Jarrar MH, Kaufmann WE. Methyl-CpG-binding protein 2 is localized in the postsynaptic compartment: an immunochemical study of subcellular fractions. Neuroscience 2003;116:77-80.
- Joyner AH, J CR, Bloss CS, Bakken TE, Rimol LM, Melle I, Agartz I, Djurovic S, Topol EJ, Schork NJ, Andreassen OA, Dale AM. A common MECP2 haplotype associates with reduced cortical surface area in humans in two independent populations. Proc Natl Acad Sci U S A 2009;106:15483-15488.
- Nagarajan RP, Hogart AR, Gwye Y, Martin MR, LaSalle JM. Reduced MeCP2 expression is frequent in autism frontal cortex and correlates with aberrant MECP2 promoter methylation. Epigenetics 2006;1:e1-e11.
- Samaco RC, Nagarajan RP, Braunschweig D, LaSalle JM. Multiple pathways regulate MeCP2 expression in normal brain development and exhibit defects in autism-spectrum disorders. Hum Mol Genet 2004;13:629-639.
- Nan X, Ng HH, Johnson CA, Laherty CD, Turner BM, Eisenman RN, Bird A. Transcriptional repression by the methyl-CpG-binding protein MeCP2 involves a histone deacetylase complex. Nature 1998;393:386-389.
- Chahrour M, Jung SY, Shaw C, Zhou X, Wong ST, Qin J, Zoghbi HY. MeCP2, a key contributor to neurological disease, activates and represses transcription. Science 2008;320:1224-1229.
- LaSalle JM. The Odyssey of MeCP2 and parental imprinting. Epigenetics 2007;2:5-10.
- Yasui DH, Peddada S, Bieda MC, Vallero RO, Hogart A, Nagarajan RP, Thatcher KN, Farnham PJ, Lasalle JM. Integrated epigenomic analyses of neuronal MeCP2 reveal a role for long-range interaction with active genes. Proc Natl Acad Sci U S A 2007;104:19416-19421.
- Amir RE, Van den Veyver IB, Wan M, Tran CQ, Francke U, Zoghbi HY. Rett syndrome is caused by mutations in X-linked MECP2, encoding methyl-CpG-binding protein 2. Nat Genet 1999;23:185-188.
- Loat CS, Curran S, Lewis CM, Duvall J, Geschwind D, Bolton P, Craig IW. Methyl-CpG-binding protein 2 polymorphisms and vulnerability to autism. Genes Brain Behav 2008;7:754-760.
- Shahbazian MD, Zoghbi HY. Molecular genetics of Rett syndrome and clinical spectrum of MECP2 mutations. Curr Opin Neurol 2001;14:171-176.
- Shibayama A, Cook EH, Feng J, Glanzmann C, Yan J, Craddock N, Jones IR, Goldman D, Heston LL, Sommer SS. MECP2 structural and 3′-UTR variants in schizophrenia, autism and other psychiatric diseases: a possible association with autism. Am J Med Genet B Neuropsychiatr Genet 2004;128B:50-53.
- Collins AL, Levenson JM, Vilaythong AP, Richman R, Armstrong DL, Noebels JL, David Sweatt J, Zoghbi HY. Mild overexpression of MeCP2 causes a progressive neurological disorder in mice. Hum Mol Genet 2004;13:2679-2689.
- Chao HT, Zoghbi HY, Rosenmund C. MeCP2 controls excitatory synaptic strength by regulating glutamatergic synapse number. Neuron 2007;56:58-65.
- Samaco RC, Fryer JD, Ren J, Fyffe S, Chao HT, Sun Y, Greer JJ, Zoghbi HY, Neul JL. A partial loss of function allele of methyl-CpG-binding protein 2 predicts a human neurodevelopmental syndrome. Hum Mol Genet 2008;17:1718-1727.
- Ramocki MB, Zoghbi HY. Failure of neuronal homeostasis results in common neuropsychiatric phenotypes. Nature 2008;455:912-918.
- Ramocki MB, Peters SU, Tavyev YJ, Zhang F, Carvalho CM, Schaaf CP, Richman R, Fang P, Glaze DG, Lupski JR, Zoghbi HY. Autism and other neuropsychiatric symptoms are prevalent in individuals with MeCP2 duplication syndrome. Ann Neurol 2009;66:771-782.
- Zhou Z, Hong EJ, Cohen S, Zhao WN, Ho HY, Schmidt L, Chen WG, Lin Y, Savner E, Griffith EC, Hu L, Steen JA, Weitz CJ, Greenberg ME. Brain-specific phosphorylation of MeCP2 regulates activity-dependent Bdnf transcription, dendritic growth, and spine maturation. Neuron 2006;52:255-269.
- Martinowich K, Hattori D, Wu H, Fouse S, He F, Hu Y, Fan G, Sun YE. DNA methylation-related chromatin remodeling in activity-dependent BDNF gene regulation. Science 2003;302:890-893.
- Rhees RW, Shryne JE, Gorski RA. Onset of the hormone-sensitive perinatal period for sexual differentiation of the sexually dimorphic nucleus of the preoptic area in female rats. J Neurobiol 1990;21:781-786.
- Arnold AP, Gorski RA. Gonadal steroid induction of structural sex differences in the central nervous system. Annu Rev Neurosci 1984;7:413-442.
- Kurian JR, Forbes-Lorman RM, Auger AP. Sex difference in mecp2 expression during a critical period of rat brain development. Epigenetics 2007;2:173-178.
- Kim KC, Choi CS, Kim JW, Han SH, Cheong JH, Ryu JH, Shin CY. MeCP2 modulates sex differences in the postsynaptic development of the valproate animal model of autism. Mol Neurobiol 2016;53:40-56.
- Carney RM, Wolpert CM, Ravan SA, Shahbazian M, Ashley-Koch A, Cuccaro ML, Vance JM, Pericak-Vance MA. Identification of MeCP2 mutations in a series of females with autistic disorder. Pediatr Neurol 2003;28:205-211.
- Beyer KS, Blasi F, Bacchelli E, Klauck SM, Maestrini E, Poustka A, International Molecular Genetic Study of Autism Consortium (IMGSAC). Mutation analysis of the coding sequence of the MECP2 gene in infantile autism. Hum Genet 2002;111:305-309.
- Lam CW, Yeung WL, Ko CH, Poon PM, Tong SF, Chan KY, Lo IF, Chan LY, Hui J, Wong V, Pang CP, Lo YM, Fok TF. Spectrum of mutations in the MECP2 gene in patients with infantile autism and Rett syndrome. J Med Genet 2000;37:E41.
- Lobo-Menendez F, Sossey-Alaoui K, Bell JM, Copeland-Yates SA, Plank SM, Sanford SO, Skinner C, Simensen RJ, Schroer RJ, Michaelis RC. Absence of MeCP2 mutations in patients from the South Carolina autism project. Am J Med Genet B Neuropsychiatr Genet 2003;117B:97-101.
- Vourc'h P, Bienvenu T, Beldjord C, Chelly J, Barthélémy C, Müh JP, Andres C. No mutations in the coding region of the Rett syndrome gene MECP2 in 59 autistic patients. Eur J Hum Genet 2001;9:556-558.
- Kurian JR, Bychowski ME, Forbes-Lorman RM, Auger CJ, Auger AP. Mecp2 organizes juvenile social behavior in a sex-specific manner. J Neurosci 2008;28:7137-7142.
- Kerr B, Alvarez-Saavedra M, Sáez MA, Saona A, Young JI. Defective body-weight regulation, motor control and abnormal social interactions in Mecp2 hypomorphic mice. Hum Mol Genet 2008;17:1707-1717.
- Cass H, Reilly S, Owen L, Wisbeach A, Weekes L, Slonims V, Wigram T, Charman T. Findings from a multidisciplinary clinical case series of females with Rett syndrome. Dev Med Child Neurol 2003;45:325-337.
- Leong WY, Lim ZH, Korzh V, Pietri T, Goh EL. Methyl-CpG binding protein 2 (Mecp2) regulates sensory function through Sema5b and Robo2. Front Cell Neurosci 2015;9:481.
- Wang C, Wang F, Li Z, Cao Q, Huang L, Chen S. MeCP2-mediated epigenetic regulation in senescent endothelial progenitor cells. Stem Cell Res Ther 2018;9:87.
- Meng X, Wang W, Lu H, He LJ, Chen W, Chao ES, Fiorotto ML, Tang B, Herrera JA, Seymour ML, Neul JL, Pereira FA, Tang J, Xue M, Zoghbi HY. Manipulations of MeCP2 in glutamatergic neurons highlight their contributions to Rett and other neurological disorders. Elife 2016;5:e14199.
- Patterson KC, Hawkins VE, Arps KM, Mulkey DK, Olsen ML. MeCP2 deficiency results in robust Rett-like behavioural and motor deficits in male and female rats. Hum Mol Genet 2016;25:3303-3320.
- Blackman MP, Djukic B, Nelson SB, Turrigiano GG. A critical and cell-autonomous role for MeCP2 in synaptic scaling up. J Neurosci 2012;32:13529-13536.
- Krämer OH, Zhu P, Ostendorff HP, Golebiewski M, Tiefenbach J, Peters MA, Brill B, Groner B, Bach I, Heinzel T, Göttlicher M. The histone deacetylase inhibitor valproic acid selectively induces proteasomal degradation of HDAC2. EMBO J 2003;22:3411-3420.
- Chen WY, Weng JH, Huang CC, Chung BC. Histone deacetylase inhibitors reduce steroidogenesis through SCF-mediated ubiquitination and degradation of steroidogenic factor 1 (NR5A1). Mol Cell Biol 2007;27:7284-7290.
- McGraw CM, Samaco RC, Zoghbi HY. Adult neural function requires MeCP2. Science 2011;333:186.
- Zhang L, He J, Jugloff DG, Eubanks JH. The MeCP2-null mouse hippocampus displays altered basal inhibitory rhythms and is prone to hyperexcitability. Hippocampus 2008;18:294-309.
- Murgatroyd C, Patchev AV, Wu Y, Micale V, Bockmühl Y, Fischer D, Holsboer F, Wotjak CT, Almeida OF, Spengler D. Dynamic DNA methylation programs persistent adverse effects of early-life stress. Nat Neurosci 2009;12:1559-1566.
- Bielsky IF, Young LJ. Oxytocin, vasopressin, and social recognition in mammals. Peptides 2004;25:1565-1574.
- Bielsky IF, Hu SB, Ren X, Terwilliger EF, Young LJ. The V1a vasopressin receptor is necessary and sufficient for normal social recognition: a gene replacement study. Neuron 2005;47:503-513.
- Veenema AH, Neumann ID. Maternal separation enhances offensive play-fighting, basal corticosterone and hypothalamic vasopressin mRNA expression in juvenile male rats. Psychoneuroendocrinology 2009;34:463-467.
- Caldwell HK, Lee HJ, Macbeth AH, Young WS. Vasopressin: behavioral roles of an “original” neuropeptide. Prog Neurobiol 2008;84:1-24.
- De Vries GJ, Simerly RB. In: Pfaff DW, Arnold AA, Etgen AM, Fahrback SE, Rubin RT. Hormones, brain and behavior. Amsterdam: Elsevier, 2002; 2002. p. 137-191.
- Auger AP, Auger CJ. In: Jirtle RL, Tyson FL. Environmental epigenomics in health and disease. Heidelberg: Springer, 2013; 2013. p. 37-54.
- Forbes-Lorman RM, Rautio JJ, Kurian JR, Auger AP, Auger CJ. Neonatal MeCP2 is important for the organization of sex differences in vasopressin expression. Epigenetics 2012;7:230-238.
- Tachibana M, Ueda J, Fukuda M, Takeda N, Ohta T, Iwanari H, Sakihama T, Kodama T, Hamakubo T, Shinkai Y. Histone methyltransferases G9a and GLP form heteromeric complexes and are both crucial for methylation of euchromatin at H3-K9. Genes Dev 2005;19:815-826.
- Anderson EM, Larson EB, Guzman D, Wissman AM, Neve RL, Nestler EJ, Self DW. Overexpression of the histone dimethyltransferase G9a in nucleus accumbens shell increases cocaine self-administration, stress-induced reinstatement, and anxiety. J Neurosci 2018;38:803-813.
- de Boer A, Vermeulen K, Egger JI, Janzing JG, de Leeuw N, Veenstra-Knol HE, den Hollander NS, van Bokhoven H, Staal W, Kleefstra T.
EHMT1 mosaicism in apparently unaffected parents is associated with autism spectrum disorder and neurocognitive dysfunction. Mol Autism 2018;9:5.
- Chase KA, Gavin DP, Guidotti A, Sharma RP. Histone methylation at H3K9: evidence for a restrictive epigenome in schizophrenia. Schizophr Res 2013;149:15-20.
- Balemans MC, Kasri NN, Kopanitsa MV, Afinowi NO, Ramakers G, Peters TA, Beynon AJ, Janssen SM, van Summeren RC, Eeftens JM, Eikelenboom N, Benevento M, Tachibana M, Shinkai Y, Kleefstra T, van Bokhoven H, Van der Zee CE. Hippocampal dysfunction in the Euchromatin histone methyltransferase 1 heterozygous knockout mouse model for Kleefstra syndrome. Hum Mol Genet 2013;22:852-866.
- Balemans MC, Ansar M, Oudakker AR, van Caam AP, Bakker B, Vitters EL, van der Kraan PM, de Bruijn DR, Janssen SM, Kuipers AJ, Huibers MM, Maliepaard EM, Walboomers XF, Benevento M, Nadif Kasri N, Kleefstra T, Zhou H, Van der Zee CE, van Bokhoven H. Reduced Euchromatin histone methyltransferase 1 causes developmental delay, hypotonia, and cranial abnormalities associated with increased bone gene expression in Kleefstra syndrome mice. Dev Biol 2014;386:395-407.
- Balemans MC, Huibers MM, Eikelenboom NW, Kuipers AJ, van Summeren RC, Pijpers MM, Tachibana M, Shinkai Y, van Bokhoven H, Van der Zee CE. Reduced exploration, increased anxiety, and altered social behavior: autistic-like features of euchromatin histone methyltransferase 1 heterozygous knockout mice. Behav Brain Res 2010;208:47-55.
- Iacono G, Dubos A, Méziane H, Benevento M, Habibi E, Mandoli A, Riet F, Selloum M, Feil R, Zhou H, Kleefstra T, Kasri NN, van Bokhoven H, Herault Y, Stunnenberg HG. Increased H3K9 methylation and impaired expression of Protocadherins are associated with the cognitive dysfunctions of the Kleefstra syndrome. Nucleic Acids Res 2018;46:4950-4965.
- Collins RE, Northrop JP, Horton JR, Lee DY, Zhang X, Stallcup MR, Cheng X. The ankyrin repeats of G9a and GLP histone methyltransferases are mono- and dimethyllysine binding modules. Nat Struct Mol Biol 2008;15:245-250.
- Rutherford LC, Nelson SB, Turrigiano GG. BDNF has opposite effects on the quantal amplitude of pyramidal neuron and interneuron excitatory synapses. Neuron 1998;21:521-530.
- Tsetsenis T, Younts TJ, Chiu CQ, Kaeser PS, Castillo PE, Südhof TC. Rab3B protein is required for long-term depression of hippocampal inhibitory synapses and for normal reversal learning. Proc Natl Acad Sci U S A 2011;108:14300-14305.
- Deguchi K, Nagamatsu G, Miyachi H, Kato Y, Morita S, Kimura H, Kitano S, Hatada I, Saga Y, Tachibana M, Shinkai Y. Posttranscriptional regulation of histone lysine methyltransferase GLP in embryonic male mouse germ cells. Biol Reprod 2013;88:36.
- Tachibana M, Nozaki M, Takeda N, Shinkai Y. Functional dynamics of H3K9 methylation during meiotic prophase progression. EMBO J 2007;26:3346-3359.
- Noh KM, Hwang JY, Follenzi A, Athanasiadou R, Miyawaki T, Greally JM, Bennett MV, Zukin RS. Repressor element-1 silencing transcription factor (REST)-dependent epigenetic remodeling is critical to ischemia-induced neuronal death. Proc Natl Acad Sci U S A 2012;109:E962-E971.
- Subbanna S, Nagre NN, Shivakumar M, Umapathy NS, Psychoyos D, Basavarajappa BS. Ethanol induced acetylation of histone at G9a exon1 and G9a-mediated histone H3 dimethylation leads to neurodegeneration in neonatal mice. Neuroscience 2014;258:422-432.
- Hiraoka M, Kagawa Y. Genetic polymorphisms and folate status. Congenit Anom (Kyoto) 2017;57:142-149.
- Jaiswal SK, Sukla KK, Chauhan A, Lakhotia AR, Kumar A, Rai AK. Choline metabolic pathway gene polymorphisms and risk for Down syndrome: an association study in a population with folate-homocysteine metabolic impairment. Eur J Clin Nutr 2017;71:45-50.
- Shaik Mohammad N, Sai Shruti P, Bharathi V, Krishna Prasad C, Hussain T, Alrokayan SA, Naik U, Radha Rama Devi A. Clinical utility of folate pathway genetic polymorphisms in the diagnosis of autism spectrum disorders. Psychiatr Genet 2016;26:281-286.
- Mitchell ES, Conus N, Kaput J. B vitamin polymorphisms and behavior: evidence of associations with neurodevelopment, depression, schizophrenia, bipolar disorder and cognitive decline. Neurosci Biobehav Rev 2014;47:307-320.
- Levav-Rabkin T, Blumkin E, Galron D, Golan HM. Sex-dependent behavioral effects of Mthfr deficiency and neonatal GABA potentiation in mice. Behav Brain Res 2011;216:505-513.
- Blumkin E, Levav-Rabkin T, Melamed O, Galron D, Golan HM. Gender-specific effect of Mthfr genotype and neonatal vigabatrin interaction on synaptic proteins in mouse cortex. Neuropsychopharmacology 2011;36:1714-1728.
- Sellers KJ, Erli F, Raval P, Watson IA, Chen D, Srivastava DP. Rapid modulation of synaptogenesis and spinogenesis by 17β-estradiol in primary cortical neurons. Front Cell Neurosci 2015;9:137.
- Brandt N, Vierk R, Rune GM. Sexual dimorphism in estrogen-induced synaptogenesis in the adult hippocampus. Int J Dev Biol 2013;57:351-356.
- Hudgens ED, Ji L, Carpenter CD, Petersen SL. The gad2 promoter is a transcriptional target of estrogen receptor (ER)alpha and ER beta: a unifying hypothesis to explain diverse effects of estradiol. J Neurosci 2009;29:8790-8797.
- Crawley JN. Behavioral phenotyping of transgenic and knockout mice: experimental design and evaluation of general health, sensory functions, motor abilities, and specific behavioral tests. Brain Res 1999;835:18-26.
- Yang M, Abrams DN, Zhang JY, Weber MD, Katz AM, Clarke AM, Silverman JL, Crawley JN. Low sociability in BTBR T+tf/J mice is independent of partner strain. Physiol Behav 2012;107:649-662.
- Schwartzer JJ, Careaga M, Onore CE, Rushakoff JA, Berman RF, Ashwood P. Maternal immune activation and strain specific interactions in the development of autism-like behaviors in mice. Transl Psychiatry 2013;3:e240.
- Defensor EB, Pearson BL, Pobbe RL, Bolivar VJ, Blanchard DC, Blanchard RJ. A novel social proximity test suggests patterns of social avoidance and gaze aversion-like behavior in BTBR T+tf/J mice. Behav Brain Res 2011;217:302-308.
- Cha SJ, Kim H, Choi HJ, Lee S, Kim K. Protein glutathionylation in the pathogenesis of neurodegenerative diseases. Oxid Med Cell Longev 2017;2017:2818565.
- Wu G, Fang YZ, Yang S, Lupton JR, Turner ND. Glutathione metabolism and its implications for health. J Nutr 2004;134:489-492.
- Pompella A, Visvikis A, Paolicchi A, De Tata V, Casini AF. The changing faces of glutathione, a cellular protagonist. Biochem Pharmacol 2003;66:1499-1503.
- Messina A, Monda V, Sessa F, Valenzano A, Salerno M, Bitetti I, Precenzano F, Marotta R, Lavano F, Lavano SM, Salerno M, Maltese A, Roccella M, Parisi L, Ferrentino RI, Tripi G, Gallai B, Cibelli G, Monda M, Messina G, Carotenuto M. Sympathetic, metabolic adaptations, and oxidative stress in autism spectrum disorders: how far from physiology. Front Physiol 2018;9:261.
- Prata J, Santos SG, Almeida MI, Coelho R, Barbosa MA. Bridging Autism Spectrum Disorders and Schizophrenia through inflammation and biomarkers - pre-clinical and clinical investigations. J Neuroinflammation 2017;14:179.
- Ming X, Johnson WG, Stenroos ES, Mars A, Lambert GH, Buyske S. Genetic variant of glutathione peroxidase 1 in autism. Brain Dev 2010;32:105-109.
- Klinga-Levan K, Andersson A, Hanson C, Ridderström M, Stenberg G, Mannervik B, Vajdy M, Szpirer J, Szpirer C, Levan G. Mapping of glutathione transferase (GST) genes in the rat. Hereditas 1993;119:285-296.
- Yochum CL, Bhattacharya P, Patti L, Mirochnitchenko O, Wagner GC. Animal model of autism using GSTM1 knockout mice and early post-natal sodium valproate treatment. Behav Brain Res 2010;210:202-210.
- Yochum CL, Bhattacharya P, Patti L, Mirochnitchenko O, Wagner GC. Animal model of autism using GSTM1 knockout mice and early post-natal sodium valproate treatment. Behav Brain Res 2010;210:202-210.
- Singhal SS, Saxena M, Ahmad H, Awasthi YC. Glutathione S-transferases of mouse liver: sex-related differences in the expression of various isozymes. Biochim Biophys Acta 1992;1116:137-146.
- Singhal SS, Saxena M, Awasthi S, Ahmad H, Sharma R, Awasthi YC. Gender related differences in the expression and characteristics of glutathione S-transferases of human colon. Biochim Biophys Acta 1992;1171:19-26.
- Stavropoulou C, Korakaki D, Rigana H, Voutsinas G, Polyzoi M, Georgakakos VN, Manola KN, Karageorgiou CE, Sambani C. Glutathione-S-transferase T1 and M1 gene polymorphisms in Greek patients with multiple sclerosis: a pilot study. Eur J Neurol 2007;14:572-574.
- Steinman KJ, Spence SJ, Ramocki MB, Proud MB, Kessler SK, Marco EJ, Green Snyder L, D'Angelo D, Chen Q, Chung WK, Sherr EH, Simons VIP Consortium. 16p11.2 deletion and duplication: characterizing neurologic phenotypes in a large clinically ascertained cohort. Am J Med Genet A 2016;170:2943-2955.
- Weiss LA, Shen Y, Korn JM, Arking DE, Miller DT, Fossdal R, Saemundsen E, Stefansson H, Ferreira MA, Green T, Platt OS, Ruderfer DM, Walsh CA, Altshuler D, Chakravarti A, Tanzi RE, Stefansson K, Santangelo SL, Gusella JF, Sklar P, Wu BL, Daly MJ, Autism Consortium. Association between microdeletion and microduplication at 16p11.2 and autism. N Engl J Med 2008;358:667-675.
- Hanson E, Bernier R, Porche K, Jackson FI, Goin-Kochel RP, Snyder LG, Snow AV, Wallace AS, Campe KL, Zhang Y, Chen Q, D'Angelo D, Moreno-De-Luca A, Orr PT, Boomer KB, Evans DW, Kanne S, Berry L, Miller FK, Olson J, Sherr E, Martin CL, Ledbetter DH, Spiro JE, Chung WK, Simons Variation in Individuals Project Consortium. The cognitive and behavioral phenotype of the 16p11.2 deletion in a clinically ascertained population. Biol Psychiatry 2015;77:785-793.
- Grissom NM, McKee SE, Schoch H, Bowman N, Havekes R, O'Brien WT, Mahrt E, Siegel S, Commons K, Portfors C, Nickl-Jockschat T, Reyes TM, Abel T. Male-specific deficits in natural reward learning in a mouse model of neurodevelopmental disorders. Mol Psychiatry 2018;23:544-555.
- Mazzucchelli C, Vantaggiato C, Ciamei A, Fasano S, Pakhotin P, Krezel W, Welzl H, Wolfer DP, Pagès G, Valverde O, Marowsky A, Porrazzo A, Orban PC, Maldonado R, Ehrengruber MU, Cestari V, Lipp HP, Chapman PF, Pouysségur J, Brambilla R. Knockout of ERK1 MAP kinase enhances synaptic plasticity in the striatum and facilitates striatal-mediated learning and memory. Neuron 2002;34:807-820.
- Lee KH, Song GJ, Kang IS, Kim SW, Paick JS, Chung CH, Rhee K. Ubiquitin-specific protease activity of USP9Y, a male infertility gene on the Y chromosome. Reprod Fertil Dev 2003;15:129-133.
- Angelakos CC, Watson AJ, O'Brien WT, Krainock KS, Nickl-Jockschat T, Abel T. Hyperactivity and male-specific sleep deficits in the 16p11.2 deletion mouse model of autism. Autism Res 2017;10:572-584.
- Courchesne E, Pierce K, Schumann CM, Redcay E, Buckwalter JA, Kennedy DP, Morgan J. Mapping early brain development in autism. Neuron 2007;56:399-413.
- Kumar VJ, Grissom NM, McKee SE, Schoch H, Bowman N, Havekes R, Kumar M, Pickup S, Poptani H, Reyes TM, Hawrylycz M, Abel T, Nickl-Jockschat T. Linking spatial gene expression patterns to sex-specific brain structural changes on a mouse model of 16p11.2 hemideletion. Transl Psychiatry 2018;8:109.
- Wilkinson B, Grepo N, Thompson BL, Kim J, Wang K, Evgrafov OV, Lu W, Knowles JA, Campbell DB. The autismassociated gene chromodomain helicase DNA-binding protein 8 (CHD8) regulates noncoding RNAs and autism-related genes. Transl Psychiatry 2015;5:e568.
- Cotney J, Muhle RA, Sanders SJ, Liu L, Willsey AJ, Niu W, Liu W, Klei L, Lei J, Yin J, Reilly SK, Tebbenkamp AT, Bichsel C, Pletikos M, Sestan N, Roeder K, State MW, Devlin B, Noonan JP. The autism-associated chromatin modifier CHD8 regulates other autism risk genes during human neurodevelopment. Nat Commun 2015;6:6404.
- Sugathan A, Biagioli M, Golzio C, Erdin S, Blumenthal I, Manavalan P, Ragavendran A, Brand H, Lucente D, Miles J, Sheridan SD, Stortchevoi A, Kellis M, Haggarty SJ, Katsanis N, Gusella JF, Talkowski ME. CHD8 regulates neurodevelopmental pathways associated with autism spectrum disorder in neural progenitors. Proc Natl Acad Sci U S A 2014;111:E4468-E4477.
- Katayama Y, Nishiyama M, Shoji H, Ohkawa Y, Kawamura A, Sato T, Suyama M, Takumi T, Miyakawa T, Nakayama KI. CHD8 haploinsufficiency results in autistic-like phenotypes in mice. Nature 2016;537:675-679.
- Suetterlin P, Hurley S, Mohan C, Riegman KL, Pagani M, Caruso A, Ellegood J, Galbusera A, Crespo-Enriquez I, Michetti C, Yee Y, Ellingford R, Brock O, Delogu A, Francis-West P, Lerch JP, Scattoni ML, Gozzi A, Fernandes C, Basson MA. Altered neocortical gene expression, brain overgrowth and functional over-connectivity in Chd8 haploinsufficient mice. Cereb Cortex 2018;28:2192-2206.
- Jung H, Park H, Choi Y, Kang H, Lee E, Kweon H, Roh JD, Ellegood J, Choi W, Kang J, Rhim I, Choi SY, Bae M, Kim SG, Lee J, Chung C, Yoo T, Park H, Kim Y, Ha S, Um SM, Mo S, Kwon Y, Mah W, Bae YC, Kim H, Lerch JP, Paik SB, Kim E. Sexually dimorphic behavior, neuronal activity, and gene expression in Chd8-mutant mice. Nat Neurosci 2018;21:1218-1228.
- Kerrisk ME, Cingolani LA, Koleske AJ. ECM receptors in neuronal structure, synaptic plasticity, and behavior. Prog Brain Res 2014;214:101-131.
- Hu VW, Frank BC, Heine S, Lee NH, Quackenbush J. Gene expression profiling of lymphoblastoid cell lines from monozygotic twins discordant in severity of autism reveals differential regulation of neurologically relevant genes. BMC Genomics 2006;7:118.
- Hu VW, Nguyen A, Kim KS, Steinberg ME, Sarachana T, Scully MA, Soldin SJ, Luu T, Lee NH. Gene expression profiling of lymphoblasts from autistic and nonaffected sib pairs: altered pathways in neuronal development and steroid biosynthesis. PLoS One 2009;4:e5775.
- Nguyen A, Rauch TA, Pfeifer GP, Hu VW. Global methylation profiling of lymphoblastoid cell lines reveals epigenetic contributions to autism spectrum disorders and a novel autism candidate gene, RORA, whose protein product is reduced in autistic brain. FASEB J 2010;24:3036-3051.
- Sato TK, Panda S, Miraglia LJ, Reyes TM, Rudic RD, Mc-Namara P, Naik KA, FitzGerald GA, Kay SA, Hogenesch JB. A functional genomics strategy reveals RORA as a component of the mammalian circadian clock. Neuron 2004;43:527-537.
- Glickman G. Circadian rhythms and sleep in children with autism. Neurosci Biobehav Rev 2010;34:755-768.
- Correia CT, Coutinho AM, Sequeira AF, Sousa IG, Lourenço Venda L, Almeida JP, Abreu RL, Lobo C, Miguel TS, Conroy J, Cochrane L, Gallagher L, Gill M, Ennis S, Oliveira GG, Vicente AM. Increased BDNF levels and NTRK2 gene association suggest a disruption of BDNF/TrkB signaling in autism. Genes Brain Behav 2010;9:841-848.
- Ylisaukko-oja T, Rehnström K, Auranen M, Vanhala R, Alen R, Kempas E, Ellonen P, Turunen JA, Makkonen I, Riikonen R, Nieminen-von Wendt T, von Wendt L, Peltonen L, Järvelä I. Analysis of four neuroligin genes as candidates for autism. Eur J Hum Genet 2005;13:1285-1292.
- Rubenstein E, Wiggins LD, Lee LC. A review of the differences in developmental, psychiatric, and medical endophenotypes between males and females with autism spectrum disorder. J Dev Phys Disabil 2015;27:119-139.
- Goodall G, Gheusi G. Abnormal patterns of maze patrolling in the mutant mouse staggerer. Behav Neural Biol 1987;47:307-320.
- Lalonde R. Exploration and spatial learning in staggerer mutant mice. J Neurogenet 1987;4:285-291.
- Lalonde R, Strazielle C. Neurobehavioral characteristics of mice with modified intermediate filament genes. Rev Neurosci 2003;14:369-385.
- Palmen SJ, van Engeland H, Hof PR, Schmitz C. Neuropathological findings in autism. Brain 2004;127:2572-2583.
- Sarachana T, Hu VW. Genome-wide identification of transcriptional targets of RORA reveals direct regulation of multiple genes associated with autism spectrum disorder. Mol Autism 2013;4:14.
- Sarachana T, Hu VW. Differential recruitment of coregulators to the RORA promoter adds another layer of complexity to gene (dys) regulation by sex hormones in autism. Mol Autism 2013;4:39.
- Sarachana T, Xu M, Wu RC, Hu VW. Sex hormones in autism: androgens and estrogens differentially and reciprocally regulate RORA, a novel candidate gene for autism. PLoS One 2011;6:e17116.
- Hu VW, Sarachana T, Sherrard RM, Kocher KM. Investigation of sex differences in the expression of RORA and its transcriptional targets in the brain as a potential contributor to the sex bias in autism. Mol Autism 2015;6:7.
- Auyeung B, Taylor K, Hackett G, Baron-Cohen S. Foetal testosterone and autistic traits in 18 to 24-month-old children. Mol Autism 2010;1:11.
- Auyeung B, Baron-Cohen S, Ashwin E, Knickmeyer R, Taylor K, Hackett G. Fetal testosterone and autistic traits. Br J Psychol 2009;100:1-22.
- Lai MC, Baron-Cohen S, Buxbaum JD. Understanding autism in the light of sex/gender. Mol Autism 2015;6:24.
- Baron-Cohen S, Auyeung B, Nørgaard-Pedersen B, Hougaard DM, Abdallah MW, Melgaard L, Cohen AS, Chakrabarti B, Ruta L, Lombardo MV. Elevated fetal steroidogenic activity in autism. Mol Psychiatry 2015;20:369-376.
- Karimi P, Kamali E, Mousavi SM, Karahmadi M. Environmental factors influencing the risk of autism. J Res Med Sci 2017;22:27.
- Kelly SJ, Ostrowski NL, Wilson MA. Gender differences in brain and behavior: hormonal and neural bases. Pharmacol Biochem Behav 1999;64:655-664.
- Savic I. Asymmetry of cerebral gray and white matter and structural volumes in relation to sex hormones and chromosomes. Front Neurosci 2014;8:329.
- Gemmel M, Hazlett M, Bögi E, De Lacalle S, Hill LA, Kokras N, Hammond GL, Dalla C, Charlier TD, Pawluski JL. Perinatal fluoxetine effects on social play, the HPA system, and hippocampal plasticity in pre-adolescent male and female rats: interactions with pre-gestational maternal stress. Psychoneuroendocrinology 2017;84:159-171.
- Morales T, Lorenson M, Walker AM, Ramos E. Both prolactin (PRL) and a molecular mimic of phosphorylated PRL, S179D-PRL, protect the hippocampus of female rats against excitotoxicity. Neuroscience 2014;258:211-217.
- Velísková J, Velísek L, Galanopoulou AS, Sperber EF. Neuroprotective effects of estrogens on hippocampal cells in adult female rats after status epilepticus. Epilepsia 2000;41:S30-S35.
- Al-Suwailem E, Abdi S, El-Ansary A. Sex differences in the glutamate signaling pathway in juvenile rats. J Neurosci Res 2018;96:459-466.
- Barth C, Villringer A, Sacher J. Sex hormones affect neurotransmitters and shape the adult female brain during hormonal transition periods. Front Neurosci 2015;9:37.
- McCarthy MM, Auger AP, Perrot-Sinal TS. Getting excited about GABA and sex differences in the brain. Trends Neurosci 2002;25:307-312.
- Kim KC, Choi CS, Kim JW, Han SH, Cheong JH, Ryu JH, Shin CY. MeCP2 modulates sex differences in the postsynaptic development of the valproate animal model of autism. Mol Neurobiol 2016;53:40-56.
- Kim KC, Cho KS, Yang SM, Gonzales EL, Valencia S, Eun PH, Choi CS, Mabunga DF, Kim JW, Noh JK, Kim HJ, Jeon SJ, Han SH, Bahn GH, Shin CY. Sex differences in autism-like behavioral phenotypes and postsynaptic receptors expression in the prefrontal cortex of TERT transgenic mice. Biomol Ther (Seoul) 2017;25:374-382.
- Robinson EB, Lichtenstein P, Anckarsäter H, Happé F, Ronald A. Examining and interpreting the female protective effect against autistic behavior. Proc Natl Acad Sci U S A 2013;110:5258-5262.