Articles

Article Tools
 View Full Text View Full Text |
 Abstract Abstract |
 Article as PDF Article as PDF |
 Print this Article Print this Article |
 Pubmed Pubmed |
 PMC PMC |
 PubReader PubReader |
 Export to Citation Export to Citation |
 Email Alerts Email Alerts |
 Open Access Open Access |
Share this article on :



Stats or Metrics
Article
Original Article
Exp Neurobiol 2018; 27(5): 408-418
Published online October 31, 2018
https://doi.org/10.5607/en.2018.27.5.408
© The Korean Society for Brain and Neural Sciences
The Novel Neuroprotective Compound KMS99220 Has an Early Anti-neuroinflammatory Effect via AMPK and HO-1, Independent of Nrf2
Ji Ae Lee1, Hye Ri Kim1, Jiyoung Kim2, Ki Duk Park3, Dong Jin Kim4, and Onyou Hwang1*
1Department of Biochemistry and Molecular Biology, University of Ulsan College of Medicine, Seoul 05505, Korea.
2Research Institute for Veterinary Science, College of Veterinary Medicine, Seoul National University, Seoul 08826, Korea.
3Convergence Research Center for Diagnosis, Treatment and Care System of Dementia, Korea Institute of Science and Technology, Seoul 02792, Korea.
4Center for Neuro-Medicine, Brain Science Institute, Korea Institute of Science and Technology, Seoul 02792, Korea.
Correspondence to: *To whom correspondence should be addressed.
TEL: 82-2-3010-4279, FAX: 82-2-3010-4248
e-mail: oyhwang@amc.seoul.kr
Abstract
We have previously reported a novel synthetic compound KMS99220 that prevented degeneration of the nigral dopaminergic neurons and the associated motor deficits, suggesting a neuroprotective therapeutic utility for Parkinson's disease. Microglia are closely associated with neuroinflammation, which plays a key role in the pathogenesis of neurodegenerative diseases. In this study, we investigated the effects of KMS99220 on the signaling involving AMP-activated protein kinase (AMPK) and heme oxygenase-1 (HO-1), the enzymes thought to regulate inflammation. KMS99220 was shown to elevate the enzyme activity of purified AMPK, and phosphorylation of cellular AMPK in BV2 microglia. It increased the level of HO-1, and this was attenuated by AMPK inhibitors. KMS99220 lowered phosphorylation of IκB, nuclear translocation of NFκB, induction of inducible nitric oxide synthase, and generation of nitric oxide in BV2 cells that had been challenged with lipopolysaccharide. This anti-inflammatory response involved HO-1, because both its pharmacological inhibition and knockdown of its expression abolished the response. The AMPK inhibitors also reversed the anti-inflammatory effects of KMS99220. The induction of HO-1 by KMS99220 occurred within 1 h, and this appeared not to involve the transcription factor Nrf2, because Nrf2 knockdown did not affect the compound's HO-1 inducing- and anti-inflammatory effects in this time window. These findings indicated that KMS99220 leads to AMPK-induced HO-1 expression in microglia, which in turn plays an important role in early anti-inflammatory signaling. Together with its neuroprotective property, KMS99220 may serve as a feasible therapeutic agent against neuroinflammation and neurodegeneration.
Graphical Abstract
Keywords: Microglia, Neuroinflammation, AMPK, HO-1, iNOS
INTRODUCTION
Neuroinflammation is mainly caused by microglia, the resident immune cells of the central nervous system. Like macrophages, the major function of microglia is to remove cell debris and pathogens in response to injury or toxic insults. Activated, inflammatory microglia are neurotoxic, as they release various neurotoxic molecules such as nitric oxide (NO), TNF-α and IL-1β, among others. If the activation status is continued due to dysfunction or aberrant activation, the consequent chronic neuroinflammation is thought to contribute to pathogenesis of neurodegenerative diseases such as Alzheimer's disease and Parkinson's disease (reviewed by [1]).
AMP-activated protein kinase (AMPK) is an enzyme involved in the regulation of cellular homeostasis and metabolic function. Accumulating evidence suggests that AMPK is also an important regulator of neuroinflammation. In microglial cells, direct pharmacological activation of AMPK lowered the lipopolysaccharide (LPS)-induced production of TNF-α, IL-6 and inducible NO synthase (iNOS) and nuclear translocation of NFκB [2,3]. In macrophages, overexpression of AMPK results in decreased inflammatory response, its knockdown leads to enhanced inflammatory response [4,5], and activation of its signaling downregulates the function of NFκB system [4,6]. Hence, AMPK is considered as a potential therapeutic target in neuroinflammation-related diseases.
The phase-2 enzyme heme oxygenase-1 (HO-1) has also been shown to possess anti-inflammatory properties. Deficiency of HO-1 exhibited abnormalities including chronic inflammation in mice [7], increased secretion of pro-inflammatory cytokines in activated mouse splenocytes [8], and hyperinflammation in human [9,10]. HO-1 induction in macrophages has been shown to mediate the switch from the proinflammatory M1 phenotype to the anti-inflammatory M2 phenotype [11]. In microglia, induction of HO-1 expression using phytochemicals or chemical agents has shown to mediate the resolution of inflammatory response [12,13,14,15].
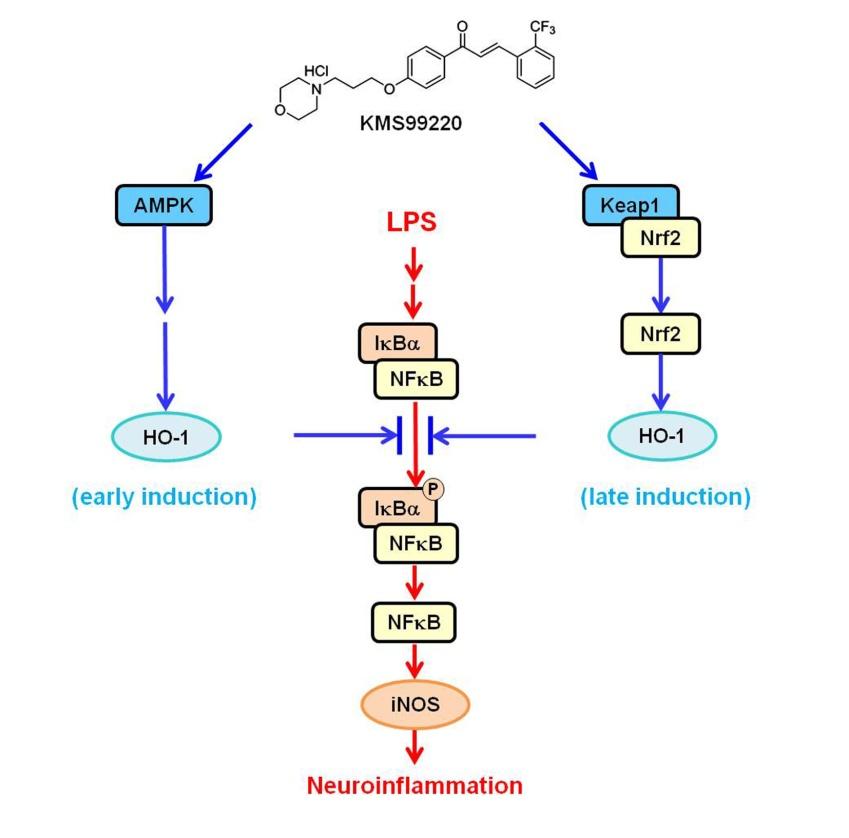
We recently synthesized a novel morpholine-containing chalcone compound KMS99220 (chemical structure shown in Fig. 7) that had a good pharmacokinetic profile and neuroprotective activity [16]. This compound exhibited excellent bioavailability and metabolic stability and no apparent side effect issues such as toxicity and cytochrome p450 inhibition. KMS99220 was shown to bind to Keap1 protein, activate Nrf2, and induce expression of its target genes including HO-1 [16]. On the other hand, it has been reported that some chalcone compounds are anti-inflammatory [17,18,19] and can activate the AMPK pathway [20,21,22,23], and that AMPK can trigger HO-1 induction [24,25,26]. Taken together, we hypothesized that KMS99220, being a chalcone, might trigger AMPK activation and HO-1 expression in microglia resulting in modulation of neuroinflammatory responses.
MATERIALS AND METHODS
Fetal bovine serum, Dulbecco's modified Eagle's medium, trypsin/EDTA, penicillin-streptomycin, and TRIzol reagent were from Thermo Fisher Scientific (Carlsbad, CA, USA). LPS, Compound C and adenine 9-β-D-arabinofuranoside (Ara A) were purchased from Sigma-Aldrich (St. Louis, MO, USA). Control small interfering RNA (siRNA), HO-1 siRNA, Nrf2 siRNA and Lipofectamine RNAiMax reagent were purchased from Thermo Fisher Scientific. Tin protoporphyrin-IX (SnPP) was from Santa Cruz Biotechnology (Santa Cruz, CA, USA). Primary antibodies used are as follows: iNOS (sc-650), lamin B (sc-6216) and HO-1 (sc-10789) from Santa Cruz Biotechnology; NFκB (NBP1-96139) from Novus Biologicals (Littleton, CO, USA); IκB (#9242), p-IκB (#2859), AMPK (#2532) and p-AMPK (#2535) from Cell Signaling Technology (Danvers, MA, USA); and β-actin (A5441) from Sigma-Aldrich. Anti-rabbit IgG, anti-goat IgG and anti-mouse IgG were purchased from Sigma-Aldrich. First Strand cDNA Synthesis kit for RT-PCR was from MBI Fermentas (Ontario, Canada). Protein concentration of samples was determined by Bradford assay (Biorad, Hempstead, UK). Enhanced luminal-based chemiluminescence western blotting detection system was obtained from Pierce Chemical (Rockford, IL, USA). CellTiter-Glo® Luminescent Cell Viability Assay kit was from Promega (Madison, WI, USA).
KMS99220 was organically synthesized according to the method we had previously published [16].
BV2 mouse microglial cells [27] were grown in Dulbecco's modified Eagle's medium with 10% fetal bovine serum in the presence of 100 IU/L penicillin and 10 µg/ml streptomycin. Nrf2sh cells and GFPsh cells were produced and grown in culture media as previously reported by us [28]. The cells were maintained at 37℃ in 95% air and 5% CO2 in humidified atmosphere.
The cell lysates and nuclear fractions were obtained according to the method previously described [29]. Protein concentrations were determined, and equal amounts (30 µg) of protein were separated on 10% SDS polyacrylamide gel and transferred onto polyvinylidene difluoride-nitrocellulose membrane. The membranes were blocked for 1 h in 20 mM Tris-HCl containing 137 mM NaCl, 0.05% Tween 20 and 5% skim milk at room temperature, incubated overnight with primary antibody against iNOS (1:200), NFκB (1:1,000), p-IκB (1:1,000), IκB (1:1,000), HO-1 (1:200), p-AMPK (1:1,000), AMPK (1:1,000), lamin B (1:200) or β-actin (1:60,000) at 4℃ followed by horseradish peroxidase-conjugated secondary antibodies for 1 h at room temperature. Protein bands were visualized using chemiluminescence substrate and quantitatively analyzed by densitometry.
The activity of AMPK enzyme was determined in the presence or absence of KMS99220 using purified AMPK protein (service provided by Eurofins Scientific, Dundee, UK). The purified AMPK was first incubated with KMS99220 in an assay buffer containing 8 mM MOPS (pH 7.0), 0.2 mM EDTA, 200 µM AMP, and 100 µM of the substrate AMARAASAAALARRR. The reaction was initiated by the addition of Mg/ATP mix (final concentrations, 10 mM magnesium acetate and 45 µM [γ-33P]-ATP). After incubation for 40 min at room temperature, the reaction was stopped by the addition of phosphoric acid (final concentration, 0.5%). Ten µl of the reaction was then spotted onto a P30 filtermat and washed four times for 4 min in 0.425% phosphoric acid and once in methanol prior to drying and scintillation counting.
Cell viability was assessed by determining the intracellular level of ATP using CellTiter-Glo® kit as described before [29].
Reverse transcription reactions were performed using 5 µg of total RNA and the First Strand cDNA Synthesis kit following the manufacturer's directions. PCR was performed at 94℃ for 30 sec, 55℃ for 40 sec, and 72℃ for 1 min for 25 cycles using specific primers: iNOS (forward, 5′-ATGTCCGAAGCAAACATCAC-3′; reverse, 5′-TAATGTCCAGGAAGTAGGTG-3′), HO-1 (forward, 5′-AGCAGGACATGGCCTCT-3′; reverse, 5′-TCTGTCAGCATCACCTGCAG-3′), NAD(P)H:quinone oxidoreductase 1 (NQO1) (forward, 5′-CCATCCTAAACAGCGATCA-3′; reverse, 5′-TAGCTTTGATCTGGTTGTC-3′), glutamate-cysteine ligase modifier subunit (GCLM) (forward, 5′-AGCTGGACTCTGTGATCATGGCTT-3′); reverse, 5′-CAAAGGCAGTCAAATCTGGTGGCA-3′), glutamate-cysteine ligase catalytic subunit (GCLC) (forward, 5′-ATGACTGTTGCCAGTGGATGAGA-3′; reverse, 5′-ACACGCCATCCTAAACAGCGATCA-3′), Nrf2 (forward, 5′-CTCGCTGGAAAAAGAAGTGG-3′; reverse, 5′-CCGTCCAGGAGTTCAGAGAG-3′), and glyceraldehydes-3-phosphate dehydrogenase (GAPDH) (forward, 5′-CACCACCATGGAGAAGGCTGG-3′; reverse,5′-TTGTCATGGATGACCTTGGCCAGG-3′).
Cells were treated with various concentrations of KMS99220 with 0.2 µg/ml LPS. After 24 h, 100 µl of culture medium was mixed with 50 µl of Griess reagent (1% sulfanilamide, 0.1% naphthylethlyene diamine dihydrochloride and 2% phosphoric acid) and incubated at room temperature for 10 min. The nitrite level was measured at 540 nm with a microplate reader (SPECTRA MAX 340 pc; Molecular Devices, Menlo Park, CA, USA).
Cells were transiently transfected with siRNA (final concentration of 40 nM) for HO-1, Nrf2 or control using Lipofectamine RNAiMax reagent according to the manufacturer's instructions. After 24 h, the cells were treated with KMS99220 or LPS, and then RT-PCR, western blot analysis and Griess assay were conducted.
Statistical tests were carried out using PRISM (GraphPad Software, San Diego, CA, USA). A value of p<0.05 was considered statistically significant. Comparisons of three or more groups were analyzed by one-way ANOVA (analysis of variance) and post Dunnett's multiple comparison tests.
RESULTS
We first examined whether our chalcone compound KMS99220 might activate AMPK. When purified AMPK protein was exposed to KMS99220, the activity of the enzyme was increased in a concentration dependent manner, with 17% elevation at 10 µM (Fig. 1A). When the murine microglial BV2 cells were exposed to KMS99220, an increase in the phosphorylated AMPK was observed in a manner dependent on KMS99220 concentration (Fig. 1B). This occurred within 15 min of the exposure, after which the level was restored to that of the untreated control (Fig. 1C). In accordance with the previous report that activated AMPK translocates into the nucleus [30], the KMS99220 treatment resulted in accumulation of the phospho-AMPK in the nucleus, between 15 and 60 min (Fig. 1D). KMS99220 had no cytotoxicity in the concentration range tested (Fig. 1E).
We tested if KMS99220 might induce HO-1 expression in microglial cells, and if so, whether this might involve AMPK. We performed RT-PCR, because we had previously demonstrated that the results of RT-PCR corresponded well to those of real-time RT-PCR for all genes investigated in the present study under our experimental conditions [18]. As shown in Fig. 2A, KMS99220 dramatically and dose-dependently elevated the mRNA and protein levels of HO-1. On the other hand, when the cells were pretreated with the AMPK pharmacological inhibitors Ara A or Compound C, the KMS99220-induced HO-1 elevation was not as apparent (Fig. 2B). This suggested that AMPK played a role in the HO-1 induction.
Since KMS99220 induced the HO-1 expression, it was possible that it might also suppress the signaling of the pro-inflammatory transcription factor NFκB. Western blot analysis showed that nuclear NFκB, which had been elevated upon exposure to the inflammagen LPS, was suppressed in the cells treated with KMS99220 (Fig. 3A). Since NFκB is known to be activated through phosphorylation and the subsequent degradation of its cytosolic inhibitor protein IκB [31], we asked if KMS99220 might also modulate this upstream step. As shown in Fig. 3B, KMS99220 indeed blocked the elevation of phospho-IκB. The decrease in the total IκB protein level after the LPS exposure, implicating degradation of phospho-IκB, was also not as apparent in the cells treated with KMS99220, supporting the notion that phosphorylation of IκB has been compromised by KMS99220.
We tested whether this effect of KMS99220 on the IκB/NFκB system might be mediated by HO-1. For this, HO-1 expression was knocked down by transfection of BV2 cells with siRNA (Fig. 3C). As shown in Fig. 3D, in the cells whose HO-1 expression was obliterated, KMS99220 was no longer able to inhibit the LPS-induced IκB phosphorylation, and there was no significant difference between the LPS-alone and the LPS+KMS99220-treated cells. This was in contrast to the cells transfected with the control siRNA, in which LPS+KMS99220 treatment lowered the level of phosphorylated IκB. This phenomenon was also confirmed using a pharmacological approach. When BV2 cells were pretreated with the HO-1 inhibitor SnPP, the inhibitory effect of KMS99220 on IκB phosphorylation was reversed, and this occurred in a dose-dependent manner (Fig. 3E).
Because activated NFκB leads to production of various proinflammatory mediators including NO in microglia, and KMS99220 was found to suppress NFκB activation, we tested whether KMS99220 might also lower NO production. For this, we assessed expression of iNOS, the NO-synthesizing enzyme, and generation of NO in LPS-activated BV2 cells. As shown in Fig. 4A, the mRNA and protein levels of iNOS, which was elevated by LPS, was dose-dependently suppressed by the co-treatment with KMS99220. The generation of NO was also suppressed: the LPS-induced NO production was dose-dependently lowered by KMS99220, and 10 µM KMS99220 was able to completely block the increase (Fig. 4B).
On the other hand, in the cells whose HO-1 expression was knocked down by transfection with HO-1 siRNA, the inhibitory effect of KMS99220 on the iNOS expression was reversed, and there was no significant difference in the iNOS level between the LPS alone and the LPS+KMS99220 treated cells (Fig. 4C). Pharmacological inhibition of HO-1 with SnPP also dose dependently alleviated the downregulating effect of KMS99220 on iNOS, completely blocking the effect at 10 µM SnPP (Fig. 4D). Taken together, the suppression of iNOS expression by KMS99220 appeared to be mediated by HO-1.
We asked whether AMPK might also play a role in the inhibitory effect of KMS99220 on the NO system. As shown in Fig 5A, pretreatement with the AMPK inhibitor Ara A was able to dose-dependently reverse the inhibitory effect of KMS99220 on the LPS-induced iNOS expression. This correlated well with the amount of NO produced under this condition (Fig. 5B). Compound C, another pharmacological inhibitor of AMPK, showed a similar effect (Fig. 5C). Taken together with the finding that these AMPK inhibitors also suppresses HO-1 induction by KMS99220 (Fig. 2), these results suggested that AMPK likely acts upstream of HO-1 in mediating the anti-inflammatory effect of KMS99220.
Since the effect of KMS99220 on the IκB/NFκB system occurred within 1 h, and HO-1 mediated this response, the HO-1 induction would be expected to be increased at an early time point. As shown in Fig. 6A, the HO-1 mRNA level was indeed elevated within 1 h of the KMS99220 treatment. As the HO-1 gene is known to be under the control of the transcription factor Nrf2, it was possible that this early induction occurred via Nrf2. However, when we examined the expression levels of NQO1, GCLM and GCLC, whose gene expressions are also known to be under the control of Nrf2 signaling, they were not changed in this time frame (Fig. 6A). This suggested that a mechanism independent of Nrf2 signaling might be involved in the KMS99220-induced early expression of HO-1.
To test this notion, we asked if the KMS99220 effect is still present in cells whose Nrf2 expression has been knocked down. For this, BV-2 cells were transfected with Nrf2 siRNA and the obliteration of Nrf2 mRNA was confirmed (Fig. 6B). When these cells were exposed to KMS99220 for only 1 h followed by a brief LPS challenge, the increase in the expression of HO-1 was evident, compared to LPS-alone control (Fig. 6C). The degree of HO-1 induction in the Nrf2 knockdown cells was not smaller than that in the control cells. This indicated that the early HO-1 induction observed after KMS99220 exposure indeed did not require Nrf2. Western blot analysis on the same samples against phosphorylated IκB revealed that the inhibition of IκB activation by KMS99220 still occurred in the absence of Nrf2 (Fig. 6C). As expected, the subsequent expression of iNOS was still downregulated in the Nrf2 knockdown cells in a manner not different from the control cells (Fig. 6D). For further confirmation, we performed the same test in BV2 microglia cells whose Nrf2 had been stably knocked down by introducing Nrf2sh [28] (Fig. 6E). Again, KMS99220 effectively induced HO-1 expression and inhibited IκB phosphorylation in the Nrf2 knockdown cells (Fig. 6F). In addition, the downregulation of iNOS expression still occurred in these cells (Fig. 6G). Taken together, these results indicated that the KMS99220-induced early induction of HO-1 and the subsequent anti-inflammatory response occurred independently of the presence of Nrf2.
DISCUSSION
With the increasing evidence that neuroinflammation plays a vital role in neurodegeneration, candidate drugs that target neuroinflammation toward therapy for neurodegenerative diseases are being actively sought. In the present study, our novel morpholinecontaining chalcone KMS99220, which was previously shown to possess a neuroprotective property with excellent pharmacological properties [16], also exerts anti-inflammatory effects on microglia, and that this is mediated by AMPK activation followed by HO-1 induction that occurs independent of the presence of Nrf2.
The present study shows that KMS99220 can suppress activation of NFκB, the inflammation-associated transcription factor, expression of its downstream gene iNOS and production of the inflammatory mediator NO. This inhibition of inflammatory signaling by KMS99220 appears to be mediated by HO-1 induction: KMS99220 is able to induce HO-1 gene expression, and the inhibitory effects of KMS99220 on iNOS expression and IκB activation are lost in both HO-1 siRNA-transfected cells and HO-1 inhibitor-treated cells. This is in line with the previous reports on the anti-inflammatory role of HO-1. The exact mechanism of anti-inflammatory activities mediated by HO-1 remains unclear, but the enzymatic metabolites bilirubin and carbon monoxide are thought to be involved. For example, it has been reported that bilirubin inhibits iNOS expression and NO production in response to endotoxin in rats [32], and that carbon monoxide attenuates NO production and NFκB activation in LPS-induced endothelial cells [33].
We show in the present study that the early induction of HO-1 expression that occurs within 1 h of KMS99220 exposure and is associated with the anti-inflammatory response might take place independent of the transcription factor Nrf2. This was demonstrated by the finding that HO-1 expression and anti-inflammatory responses still occur in the absence of Nrf2. In addition, the expression levels of other Nrf2-dependent genes, such as NQO1, GCLM and GCLC, were not increased in this time frame. We show that the early expression of HO-1 is dependent on AMPK signaling. KMS99220 was able to activate AMPK within 15 min and induce HO-1 expression, and inhibition of AMPK activity abolished the HO-1 induction. Other transcription factors reported to be involved in HO-1 induction include CREB [34,35] and FOXO1 [36,37], and they are reported to be activated by AMPK [38,39]. Therefore, it is possible to speculate that AMPK, upon activation by KMS99220 and subsequent translocation into the nucleus, acts on any of these transcription factors and leads to the early HO-1 expression.
We have previously shown that KMS99220 is able to bind to Keap1, the inhibitory protein for Nrf2, and lead to Nrf2 activation [16]. Because HO-1 is a target gene of Nrf2, it was possible that the KMS99220-induced HO-1 elevation occurred via this signaling. However, we noted a discrepancy in time course, in that the suppression of IκB phosphorylation and NFκB nuclear localization by KMS99220 took place faster than the increase in HO-1 resulting from Nrf2 activation. This led us to ask whether there might be another, earlier mechanism for activation of the anti-inflammatory response. The present study shows that AMPK activation and the resulting expression of HO-1 occurs faster, within the same time window as the NFκB activation. Since KMS99220 also interacts with Keap1, it can be postulated that the KMS99220-induced HO-1 induction is biphasic, first via AMPK activation and later via Nrf2 signaling (Fig. 7).
KMS99220 appears to act as direct activator of AMPK, because it led to increased enzyme activity of purified AMPK protein. Studies have suggested the utility of AMPK-activating compounds as a therapeutic agent for neuroinflammatory disorders. In microglial cells, pharmacological activation of AMPK using its direct AMPK agonists 5-amino-4-imidazole carboxamide riboside and ENERGI-F704 lowered the LPS-induced production of TNF-α, IL-6 and iNOS and nuclear translocation of NFκB [2,3]. In addition, activation of AMPK resulting from exposure to the phytochemicals such as (+)catechin, resveratrol, and lycopene have all been linked to suppression of microglial activation [13,40,41]. In macrophages, overexpression of AMPK results in decreased production of TNF-α and IL-6 after LPS exposure, whereas knockdown of AMPK expression leads to increased production of these proinflammatory cytokines [4]. In addition, macrophages generated from AMPKα1-deficient mice exhibited enhanced inflammatory response [5], and the AMPK signaling downregulates the function of NFκB system [4,6].
In conclusion, our novel chalcone compound KMS99220 activates AMPK signaling, leading to downregulation of inflammatory response in microglia, and this appears to be mediated by HO-1 that is induced Nrf2-indepdendently downstream of AMPK at an earlier time point. Together with our previous finding that KMS99220 exhibits excellent neuroprotective property and pharmacokinetic profile, the compound might be utilized as a potential candidate for therapy of neuroinflammation- and neurodegeneration-associated disorders.
Figures

{kind=link}

{kind=link}

{kind=link}

{kind=link}

{kind=link}

{kind=link}
{kind=link}
References
- Labzin LI, Heneka MT, Latz E. Innate immunity and neurodegeneration. Annu Rev Med 2018;69:437-449.

- Chen CC, Lin JT, Cheng YF, Kuo CY, Huang CF, Kao SH, Liang YJ, Cheng CY, Chen HM. Amelioration of LPS-induced inflammation response in microglia by AMPK activation. BioMed Res Int 2014;2014:692061.
- Giri S, Nath N, Smith B, Viollet B, Singh AK, Singh I. 5-aminoimidazole-4-carboxamide-1-beta-4-ribofuranoside inhibits proinflammatory response in glial cells: a possible role of AMP-activated protein kinase. J Neurosci 2004;24:479-487.
- Sag D, Carling D, Stout RD, Suttles J. Adenosine 5′-monophosphate-activated protein kinase promotes macrophage polarization to an anti-inflammatory functional phenotype. J Immunol 2008;181:8633-8641.
- Carroll KC, Viollet B, Suttles J. AMPKα1 deficiency amplifies proinflammatory myeloid APC activity and CD40 signaling. J Leukoc Biol 2013;94:1113-1121.
- Yang Z, Kahn BB, Shi H, Xue BZ. Macrophage alpha1 AMP-activated protein kinase (alpha1AMPK) antagonizes fatty acid-induced inflammation through SIRT1. J Biol Chem 2010;285:19051-19059.
- Poss KD, Tonegawa S. Heme oxygenase 1 is required for mammalian iron reutilization. Proc Natl Acad Sci U S A 1997;94:10919-10924.
- Kapturczak MH, Wasserfall C, Brusko T, Campbell-Thompson M, Ellis TM, Atkinson MA, Agarwal A. Heme oxygenase-1 modulates early inflammatory responses: evidence from the heme oxygenase-1-deficient mouse. Am J Pathol 2004;165:1045-1053.
- Greil J, Verga-Falzacappa MV, Echner NE, Behnisch W, Bandapalli OR, Pechanska P, Immenschuh S, Vijayan V, Balla J, Tsukahara H, Schneider M, Janka G, Claus M, Longerich T, Muckenthaler MU, Kulozik AE. Mutating heme oxygenase-1 into a peroxidase causes a defect in bilirubin synthesis associated with microcytic anemia and severe hyperinflammation. Haematologica 2016;101:e436-e439.
- Radhakrishnan N, Yadav SP, Sachdeva A, Pruthi PK, Sawhney S, Piplani T, Wada T, Yachie A. Human heme oxygenase-1 deficiency presenting with hemolysis, nephritis, and asplenia. J Pediatr Hematol Oncol 2011;33:74-78.
- Weis N, Weigert A, von Knethen A, Brüne B. Heme oxygenase-1 contributes to an alternative macrophage activation profile induced by apoptotic cell supernatants. Mol Biol Cell 2009;20:1280-1288.
- Lee EJ, Ko HM, Jeong YH, Park EM, Kim HS. β-Lapachone suppresses neuroinflammation by modulating the expression of cytokines and matrix metalloproteinases in activated microglia. J Neuroinflammation 2015;12:133.
- Lin HY, Huang BR, Yeh WL, Lee CH, Huang SS, Lai CH, Lin H, Lu DY. Antineuroinflammatory effects of lycopene via activation of adenosine monophosphate-activated protein kinase-α1/heme oxygenase-1 pathways. Neurobiol Aging 2014;35:191-202.
- Lee DS, Jeong GS. Butein provides neuroprotective and anti-neuroinflammatory effects through Nrf2/ARE-dependent haem oxygenase 1 expression by activating the PI3K/Akt pathway. Br J Pharmacol 2016;173:2894-2909.
- Sun GY, Chen Z, Jasmer KJ, Chuang DY, Gu Z, Hannink M, Simonyi A. Quercetin attenuates inflammatory responses in BV-2 microglial cells: role of MAPKs on the Nrf2 pathway and induction of heme oxygenase-1. PLoS One 2015;10:e0141509.
- Lee JA, Son HJ, Choi JW, Kim J, Han SH, Shin N, Kim JH, Kim SJ, Heo JY, Kim DJ, Park KD, Hwang O. Activation of the Nrf2 signaling pathway and neuroprotection of nigral dopaminergic neurons by a novel synthetic compound KMS99220. Neurochem Int 2018;112:96-107.
- Mahapatra DK, Bharti SK, Asati V. Chalcone derivatives: anti-inflammatory potential and molecular targets perspectives. Curr Top Med Chem 2017;17:3146-3169.
- Lee JA, Kim JH, Woo SY, Son HJ, Han SH, Jang BK, Choi JW, Kim DJ, Park KD, Hwang O. A novel compound VSC2 has anti-inflammatory and antioxidant properties in microglia and in Parkinson's disease animal model. Br J Pharmacol 2015;172:1087-1100.
- Wu J, Li J, Cai Y, Pan Y, Ye F, Zhang Y, Zhao Y, Yang S, Li X, Liang G. Evaluation and discovery of novel synthetic chalcone derivatives as anti-inflammatory agents. J Med Chem 2011;54:8110-8123.
- Zhang X, Zhu P, Zhang X, Ma Y, Li W, Chen JM, Guo HM, Bucala R, Zhuang J, Li J. Natural antioxidant-isoliquiritigenin ameliorates contractile dysfunction of hypoxic cardiomyocytes via AMPK signaling pathway. Mediators Inflamm 2013;2013:390890.
- Zhang T, Yamamoto N, Ashida H. Chalcones suppress fatty acid-induced lipid accumulation through a LKB1/AMPK signaling pathway in HepG2 cells. Food Funct 2014;5:1134-1141.
- Han JY, Park SH, Yang JH, Kim MG, Cho SS, Yoon G, Cheon SH, Ki SH. Licochalcone suppresses LXRα-induced hepatic lipogenic gene expression through AMPK/Sirt1 pathway activation. Toxicol Res 2014;30:19-25.
- Dang Y, Ling S, Duan J, Ma J, Ni R, Xu JW. Bavachalcone-induced manganese superoxide dismutase expression through the AMP-activated protein kinase pathway in human endothelial cells. Pharmacology 2015;95:105-110.
- Liu XM, Peyton KJ, Shebib AR, Wang H, Korthuis RJ, Durante W. Activation of AMPK stimulates heme oxygenase-1 gene expression and human endothelial cell survival. Am J Physiol Heart Circ Physiol 2011;300:H84-H93.
- Zimmermann K, Baldinger J, Mayerhofer B, Atanasov AG, Dirsch VM, Heiss EH. Activated AMPK boosts the Nrf2/HO-1 signaling axis--A role for the unfolded protein response. Free Radic Biol Med 2015;88:417-426.
- Gao XY, Wang SN, Yang XH, Lan WJ, Chen ZW, Chen JK, Xie JH, Han YF, Pi RB, Yang XB. Gartanin protects neurons against glutamate-induced cell death in HT22 cells: independence of Nrf-2 but involvement of HO-1 and AMPK. Neurochem Res 2016;41:2267-2277.
- Blasi E, Barluzzi R, Bocchini V, Mazzolla R, Bistoni F. Immortalization of murine microglial cells by a v-raf/v-myc carrying retrovirus. J Neuroimmunol 1990;27:229-237.
- Koh K, Cha Y, Kim S, Kim J. tBHQ inhibits LPS-induced microglial activation via Nrf2-mediated suppression of p38 phosphorylation. Biochem Biophys Res Commun 2009;380:449-453.
- Woo SY, Kim JH, Moon MK, Han SH, Yeon SK, Choi JW, Jang BK, Song HJ, Kang YG, Kim JW, Lee J, Kim DJ, Hwang O, Park KD. Discovery of vinyl sulfones as a novel class of neuroprotective agents toward Parkinson's disease therapy. J Med Chem 2014;57:1473-1487.
- Zaha VG, Young LH. AMP-activated protein kinase regulation and biological actions in the heart. Circ Res 2012;111:800-814.
- Kawai T, Akira S. Signaling to NF-kappaB by Toll-like receptors. Trends Mol Med 2007;13:460-469.
- Wang WW, Smith DL, Zucker SD. Bilirubin inhibits iNOS expression and NO production in response to endotoxin in rats. Hepatology 2004;40:424-433.
- Sun B, Zou X, Chen Y, Zhang P, Shi G. Preconditioning of carbon monoxide releasing molecule-derived CO attenuates LPS-induced activation of HUVEC. Int J Biol Sci 2008;4:270-278.
- Barnett M, Hall S, Dixit M, Arany I. Simvastatin attenuates oleic acid-induced oxidative stress through CREB-dependent induction of heme oxygenase-1 in renal proximal tubule cells. Pediatr Res 2016;79:243-250.
- Astort F, Repetto EM, Rocha-Viegas L, Mercau ME, Puch SS, Finkielstein CV, Pecci A, Cymeryng CB. Role of CREB on heme oxygenase-1 induction in adrenal cells: involvement of the PI3K pathway. J Mol Endocrinol 2016;57:113-124.
- Liu X, Cui Y, Li M, Xu H, Zuo J, Fang F, Chang Y. Cobalt protoporphyrin induces HO-1 expression mediated partially by FOXO1 and reduces mitochondria-derived reactive oxygen species production. PLoS One 2013;8:e80521.
- Kang J, Jeong MG, Oh S, Jang EJ, Kim HK, Hwang ES. A FoxO1-dependent, but NRF2-independent induction of heme oxygenase-1 during muscle atrophy. FEBS Lett 2014;588:79-85.
- Thomson DM, Herway ST, Fillmore N, Kim H, Brown JD, Barrow JR, Winder WW. AMP-activated protein kinase phosphorylates transcription factors of the CREB family. J Appl Physiol (1985) 2008;104:429-438.
- Kim TT, Dyck JR. Is AMPK the savior of the failing heart?. Trends Endocrinol Metab 2015;26:40-48.
- Syed Hussein SS, Kamarudin MN, Kadir HA. (+)-Catechin attenuates NF-κB activation through regulation of Akt, MAPK, and AMPK signaling pathways in LPS-induced BV-2 microglial cells. Am J Chin Med 2015;43:927-952.
- Han Y, Jiang C, Tang J, Wang C, Wu P, Zhang G, Liu W, Jamangulova N, Wu X, Song X. Resveratrol reduces morphine tolerance by inhibiting microglial activation via AMPK signalling. Eur J Pain 2014;18:1458-1470.