Articles

Article Tools
 View Full Text View Full Text |
 Abstract Abstract |
 Article as PDF Article as PDF |
 Print this Article Print this Article |
 Pubmed Pubmed |
 PMC PMC |
 PubReader PubReader |
 Export to Citation Export to Citation |
 Email Alerts Email Alerts |
 Open Access Open Access |
Share this article on :



Stats or Metrics
Article
Original Article
Exp Neurobiol 2019; 28(1): 85-103
Published online January 21, 2019
https://doi.org/10.5607/en.2019.28.1.85
© The Korean Society for Brain and Neural Sciences
Restorative Mechanism of Neural Progenitor Cells Overexpressing Arginine Decarboxylase Genes Following Ischemic Injury
Jae Young Kim1, Jong Youl Kim1, Jae Hwan Kim1,4, Hosung Jung1,2, Won Taek Lee1, and Jong Eun Lee1,2,3*
1Department of Anatomy, Yonsei University College of Medicine, Seoul 03722, Korea.
2BK21 PLUS Project for Medical Science, Yonsei University College of Medicine, Seoul 03722, Korea.
3Brain Research Institute, Yonsei University College of Medicine, Seoul 03722, Korea.
4Center for Neuroscience Imaging Research (CNIR), Institute for Basic Science, Sungkyunkwan University, Suwon 16419, Korea.
Correspondence to: *To whom correspondence should be addressed.
TEL: 82-2-2228-1646, FAX: 82-2-365-0700
e-mail: jelee@yuhs.ac
Abstract
Cell replacement therapy using neural progenitor cells (NPCs) following ischemic stroke is a promising potential therapeutic strategy, but lacks efficacy for human central nervous system (CNS) therapeutics. In a previous
Graphical Abstract
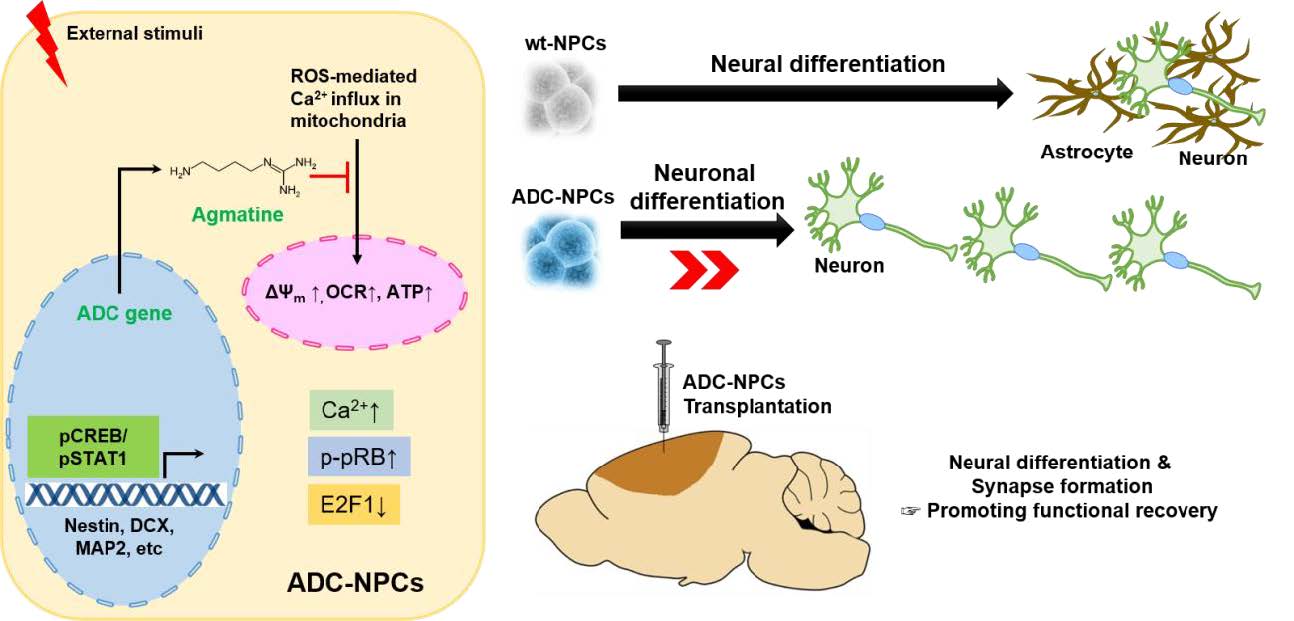
Keywords: Ischemic stroke, Cell replacement therapy, Neural progenitor cells, Arginine decarboxylase, Cell cycle arrest, Neural differentiation
INTRODUCTION
Neural progenitor cells (NPCs) are promising candidates for cell transplantation to treat pathophysiological conditions of the central nervous system (CNS) such as Alzheimer disease, multiple sclerosis, hypoxic ischemic encephalopathy, and ischemic stroke [1,2,3,4,5,6]. However, the clinical application of stem cell technology faces several technical and safety challenges, particularly heterogeneity, poor neuronal yield, and low survival rate. Neurodegenerative diseases–including ischemic stroke–are characterized by irreversible tissue damage resulting from hemodynamic, cellular, and molecular changes [7,8], which affect both the resident cells and the therapeutically transplanted NPCs. One of the prominent molecular changes accompanying neurodegenerative diseases is an increase in the level of reactive oxygen species (ROS), which causes the oxidization of cellular components, excessive calcium influx, the breakdown of mitochondrial membrane potential, and cell death [8,9]. Recently, it has become clear that balanced regulation of ROS is of critical significance for cell fate determination as well as for stem cell differentiation, function, and survival following transplantation in injured tissues [10]. Therefore, the development of advanced cell replacement therapies for neurodegenerative diseases is required, and the development of robust NPCs capable of surviving in injured environments is essential. Agmatine (NH2[CH2]4NH2C[NH]=NH; PubChem CID: 199), the natural metabolite of arginine, interacts with several intracellular molecular targets before being exported out of the cell [11], and modulates neuronal functions in
MATERIALS AND METHODS
For the
We cultured the NPCs as described previously [15]. Briefly, we extracted E14.5 embryos (Koatech Technology Corp., Republic of Korea) from placental tissue under a surgical microscope using surgical kits. The striatum was removed from the fetal brain and placed in Hank's balanced salt solution (HBSS; Thermo Scientific, USA). The dissected tissues were allowed to settle for 3 min, then centrifuged at 8×g for 5 min. The tissue pellets were gently triturated by repeated passage through a fire-polished, constricted Pasteur pipette in murine NeuroCult™ NSC basal medium containing a proliferation supplement (Stem Cell Technologies, USA) and 20 ng/ml epidermal growth factor (EGF) (Invitrogen, USA). The cells were counted and plated into T25 flasks at a density of 2.5~3×106 cells/10 ml. The culture flasks were incubated in a humidified atmosphere comprising 95% air and 5% CO2 at 37℃. We replaced the culture media every 3 days. We used the striatal NPCs for the
The full-length human arginine decarboxylase (ADC) gene complementary DNA (cDNA) (GenBank accession number: AY325129.1) was constructed as described previously [14,15,16,20]. We filtered the highest titer of retrovirus-containing medium (
OGD was applied as previously described [21,22]. Briefly, the NSC media were replaced with filtered, deoxygenated, glucose-free Eagle's balanced salt solution (BSS0.0). The NPCs were placed in an anaerobic chamber (Coy Laboratories, Grass Lake, MI, USA) containing a gas mixture comprising 5% CO2, 5% H2, and 85% N2 (0.2% O2) under glucose-free anaerobic conditions at 37℃ for 2 h. OGD was terminated by returning the cells to normoxic conditions in normal NSC (proliferation) medium containing a proliferation supplement and 20 ng/ml EGF.
We harvested the NPCs by centrifugation (12,000×
We performed western blot analysis using equal amounts of protein (15 mg) of whole-cell, cytoplasmic, and nuclear extraction (NE-PER 78835, Thermo Scientific, USA), and striatal tissue lysates prepared from the indicated groups, as described previously [15]. We blocked the membranes for 1 h at room temperature with 5% skim milk or bovine serum albumin in 1×Tris-buffered saline (TBS) and 0.1% Tween-20 (TBST). We incubated the membranes overnight at 4℃ with the following primary antibodies: anti-arginine decarboxylase (ADC) (ADC, 1:500; ATGEN), anti-doublecortin (DCX) (AB23542, 1:1000; ABCAM), anti-GFAP (MS1376, 1:200; Thermo Scientific), anti-Olig2 (AB81093, 1:1000; ABCAM), anti-cyclin-dependent kinase 4 (CDK4) (SC601, 1:1000; Santa Cruz), anti-phospho-retinoblastoma protein (p-pRB Ser 807/811) (#9308, 1:1000; Cell Signaling), anti-retinoblastoma protein (pRB) (SC50, 1:1000; Santa Cruz), anti-E2F1 (SC193, 1:1000; Santa Cruz), anti-phospho-p44/42 MAPK (pERK1/2 Thr202/Tyr204) (#9101, 1:1000; Cell Signaling), anti-p44/42 MAPK (ERK1/2) (#9102, 1:1000; Cell Signaling), anti-phospho-CREB (pCREB Ser133) (#9198, 1:1000; Cell Signaling), anti-CREB (#9197, 1:1000, Cell Signaling), anti-phospho-STAT1 (pSTAT1 Ser727) (#9177, 1:1000; Cell Signaling), anti-STAT1 (#9172, 1:1000; Cell Signaling), anti-synaptophysin (MAB368, 1:1000; Millipore), anti-GAP43 (#8945, 1:1000; Cell Signaling), anti-PSD95 (AB18258, 1:1000; ABCAM), anti-neurofilament (04-1032, 1:1000; Millipore), anti-beta-actin (AB8226, 1:1000; ABCAM), and anti-lamin-B1 (SC6217, 1:1000; Santa Cruz). We washed the membranes three times with TBST every 10 mins, and incubated them with secondary antibodies conjugated to horseradish peroxidase in TBST for 1 h at room temperature. Finally, we rinsed the membranes and visualized the proteins using a SuperSignal West Pico Chemiluminescent Substrate protein detection kit according to the manufacturer's instructions. We visualized the blots using LAS 4000 mini image analysis software (GE Healthcare, Buckinghamshire, UK), and carried out densitometric measurements on all the detected proteins using Image J.
We carried out immunochemistry with the indicated groups as previously described [15]. We washed the NPCs and brain tissues three times with PBS, fixed them with 4% paraformaldehyde for 3 h, and washed them again with PBS. We permeabilized the NPCs with PBS containing 0.025% Triton X-100 (PBST), and blocked them for 1 h at room temperature with goat-blocking serum (Invitrogen) or 1% bovine serum albumin in PBS. We incubated the NPCs overnight at 4℃ with the following primary antibodies in antibody diluent solution (Invitrogen): anti-arginine decarboxylase (ADC) (ADC, 1:200; ATGEN), anti-agmatine (AB1568, 1:200; Chemicon), anti-nestin (MAB353, 1:200; Millipore), anti-doublecortin (DCX) (AB23542, 1:200; ABCAM), anti-S100β (AB52642, 1:200; ABCAM), anti-GFAP (MS1376, 1:200; Thermo Scientific), anti-Olig2 (AB81093, 1:200; ABCAM), anti-myelin basic protein (MBP) (AB7349, 1:200; ABCAM), anti-cyclin-dependent kinase 4 (CDK4) (SC601, 1:200; Santa Cruz), anti-phospho-retinoblastoma protein (p-pRB Ser 807/811) (#9308, 1:200; Cell Signaling), anti-retinoblastoma protein (pRB) (SC50, 1:200; Santa Cruz), anti-E2F1 (SC193, 1:200; Santa Cruz), anti-phospho-p44/42 MAPK (pERK1/2 Thr202/Tyr204) (#9101, 1:200; Cell Signaling), anti-phospho-CREB (pCREB Ser133) (#9198, 1:200; Cell Signaling), anti-phospho-STAT1 (pSTAT1 Ser727) (#9177, 1:200; Cell Signaling), anti-MAP2 (AB5392, 1:200; ABCAM), anti-synaptophysin (MAB368, 1:200; Millipore), and anti-PSD95 (AB18258, 1:200; ABCAM). We removed the primary antibodies and washed the NPCs three times for 3 min each time with PBST. We then incubated the samples with the appropriate secondary antibodies conjugated to FITC, rhodamine, and Alexa Fluor® 405 prepared in fluorescent antibody diluent solution (1:200~1:500; ABCAM) for 2 h at room temperature. We washed the NPCs again three times for 3 mins each time with PBST. Finally, we counterstained the NPCs with 4′,6-diamidino-2-phenylindole (DAPI) and visualized them using an LSM 710 confocal microscope.
Double-stranded siRNAs (21-mer, AccuTarget™ genome-wide predesigned siRNAs) targeting
We chemically dissociated the NPCs into single cells and attached them to a glass bottom dish (SPL, Republic of Korea) coated with poly-L-ornithine (P4957, Sigma) for 1 h. NPCs were loaded with the Ca2+-sensitive green fluorescence indicator Fluo-4 AM (4 µM, F14217; Molecular Probes®) diluted in Tyrode's buffer (2Ca5K solution (319 mOsm) containing 2 mM CaCl2, 1 mM MgCl2, 140 mM NaCl, 5 mM KCl, 10 mM 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES) buffer (pH 7.4, Sigma), 10 mM glucose, and 0.5 M ethylenediaminetetraacetic acid (EDTA) at 37℃ for 30 min. We prepared zero-calcium solutions by replacing CaCl2 with equimolar MgCl2 and 0.5 mM EDTA [24]. We examined the Fluo-4AM-labeled Ca2+ using an LSM 710 confocal microscope. The Fluo-4AM signal was recorded every 3 sec for 5 min, and the signal intensity (region of interest; ROI) was calculated using ZEN 2011 measurement software. The Fluo-4AM intensity was calculated using the following calibration equation [25]:

Where kd is the dissociation constant of the indicator (Fluo-4AM; 345 nM).
We used a Seahorse XF24 Extracellular Flux Analyzer (Seahorse Biosciences; North Billerica, MA, USA) to measure the oxygen consumption rate (OCR) in the cells. We seeded the dissociated NPCs onto 24-well Seahorse microplates at a density of 1.5×105 cells per well. Before taking the measurements, we soaked the sensor cartridges in XF Calibrant Solution (Seahorse Biosciences) in 24-well cell culture microplates overnight at 37℃ to hydrate them. We filled the injection ports on the sensor cartridge with drugs from an XF Cell Mito Stress Test Kit (Seahorse Bioscience): oligomycin (1 µM), carbonyl cyanide 4-(trifluoromethoxy) phenylhydrazone (FCCP) (1 µM), and rotenone (1 µM) with antimycin A (1 µM). The XF24 microplate was loaded into the Seahorse XF24 analyzer according to the manufacturer's instructions. All experiments were carried out at 37℃.
We carried out transient middle cerebral artery occlusion (tMCAO) as previously described [18]. In brief, we intramuscularly anesthetized Sprague Dawley rats with Zoletil 50 (0.6 mg/kg; Virbac) and Rompun (0.4 mg/kg). We monitored the body temperatures of the animals with a rectal probe and ensured they remained at between 36.5℃ and 37.5℃ using heating pads. We anesthetized all the experimental animals before surgery. We then exposed the right common carotid artery (rCCA), the external carotid artery (ECA), and the internal carotid artery (ICA) of each rat through a ventral midline incision. We introduced a 4–0 monofilament nylon suture into the CCA lumen of each rat and gently advanced through the ICA to the MCA until the regional cerebral blood flow was reduced to 15~20% of baseline as monitored by a laser Doppler flow meter (OMEGA FLOW, FLO-C1, Neuroscience, Tokyo, Japan). After 1 h of occlusion, we withdrew the suture to restore blood flow. We kept the animals on the heating pad and allowed them
We transplanted the ADC-NPCs (1×106 cells/10 µl) into the ipsilateral striatum (posterior +0.3 mm, lateral 3 mm, ventral 5 mm from bregma) using a 25 G Hamilton syringe 7 days after MCAO using a stereotaxic apparatus. After cell transplantation, we placed the needle into the injection site over a 10 min period to inhibit backflow.
We decapitated the rats after ischemia, and rapidly removed the brains and coronal sections obtained at 2-mm intervals. We measured the volume of the infarction in each animal as the slice thickness (2 mm), and calculated the sum of the infarction areas in all the brain slices. The brain infarct volume was determined using the following formula: brain infarct volume (%)=(the volume of the ipsilateral hemisphere/the volume of the contralateral hemisphere) ×100.
After MCAO, we assessed the behavior of each rat using the rotarod test to detect improvements in neurological motor function. We increased the rotarod (Med Associates Inc.) speed from 4 to 40 rpm. We tested the rats five times on each testing day allowing them a minimum of 5 min rest in the home cage between trials.
We performed automatic colocalization assays for the fluorescence images using the Pearson's correlation coefficient (PCC) and Manders' overlap coefficients (MOCs) K1 & K2 using the JACoP plugin for ImageJ [26,27,28,29]. Briefly, the Pearson's coefficient provides an estimate of the goodness of fit and the rate of association between two fluorochromes. Its value can range from 1 to −1: 1 represents complete positive correlation; −1 represents negative correlation; 0 represents no correlation; 0.8≥r≥0.5 indicates a moderate positive value of colocalization; and 0.5≥r≥0 indicates a weak positive value of colocalization. In the present study, the PCC represented the value of colocalization in the nucleus, analyzed using a specific antigen (channel 1) and DAPI (channel 2). The MOC is calculated as the Pearson's coefficient with the mean intensity value of both channels taken out of the expression; its value is defined as between 0 and 1. Two additional overlap coefficients, K1 & K2, were used to split the mean value of colocalization: the former is related to the first channel total intensity, the latter is related to the second channel total intensity. In the present study, MOC K1 & K2 are represented as the single fluorescence intensity in the merged figure, analyzed with a specific antigen (channel 1: K1 and channel 2: K1+K2). To determine the single fluorescence intensity in the merged figure, all MOC graphs represent the K1 value.
We carried out statistical tests to determine differences between groups using the Student's
RESULTS
We subjected the ADC-NPCs to OGD for 2 h following ischemic stress to investigate their therapeutic potential. ADC protein and agmatine levels both increased significantly in the ADC-NPCs under normal and OGD conditions, but the expression of ADC protein was higher under OGD conditions than under normal conditions (Fig. 1A and 1B). During ischemic stress, ADC protein was also expressed in the wild-type NPCs (wt-NPCs), but the expression level was lower than in the ADC-NPCs (Fig. 1A and 1B). The number of PI-positive ADC-NPCs decreased significantly following OGD (Fig. 1C and 1D). To identify cell cycle regulation in the wt-NPCs and ADC-NPCs, we subjected the PI-stained cells to flow cytometry at DIV7 under both normal and OGD conditions. The FACS results indicated that ADC-NPCs significantly reduced the cell population of G0-G1 phase (77.31±2.21% of wt-NPCs vs. 61.68±5.93% of ADC-NPCs) and extended the cell population of G2-M phase (19.17±2.12% of wt-NPCs vs. 34.31±5.47% of ADC-NPCs) following normal condition. In contrast, ADC-NPCs significantly extended the cell population of G0–G1 phase (74.54±1.3% of wt-NPCs vs. 82.25±0.9% of ADC-NPCs) following OGD, suggesting that the ADC-NPCs induced a shift in the cell cycle from proliferation to quiescence/differentiation in the G0-G1 phase following OGD injury (Fig. 1E). These results indicate that ADC and agmatine overexpression in ADC-NPCs attenuate cell death and cell cycle exit following ischemic stress. It has been reported that NPCs differentiate into different neuronal cells in a suitable environment. With this view to investigating the neural differentiation of ADC-NPCs during ischemic stress, we investigated the expression levels of various proteins in ADC-NPCs subjected to OGD. Both western blot and immunostaining analyses revealed that the expression levels of neuronal marker DCX, astroglial marker GFAP, and oligodendroglial marker Olig2 increased in the ADC-NPCs under normal conditions (Fig. 2A~2E). However, the expression levels of DCX and Olig2 were four times greater in the ADC-NPCs than in the normal NPCs following OGD injury, and the expression of GFAP was significantly lower in the ADC-NPCs after the same insult (Fig. 2A~2C and 2E). These results suggest that the overexpression of ADC genes in NPCs attenuates cell death, cell cycle exit, and enhanced neural differentiation following ischemic stress.
Cell cycle progression is an important determinant of the developmental process. Cellular differentiation in multicellular organisms can be initiated through cell cycle withdrawal via transcriptional regulation [30]. To identify cell cycle regulation in the wt-NPCs and ADC-NPCs, we investigated some of the essential components of the G1-S phase transition in eukaryotic cells, i.e., CDK4/2, pRB, p-pRB, and E2F1/DP-1. The CDK4 complex (CDK4/6-cyclin D) and the CDK2 complex (CDK2-cyclin E) sequentially phosphorylate retinoblastoma protein (pRB), which possible to bind the transcription complex E2F-DP1. In an uncommitted cell, hypo-phosphorylated pRB hinder to bind the E2F1-DP1 transcription factor in the S phase-specific genes, suggesting that the pRB phosphorylation is essential to enter the G1-S transition [31,32].
In the present study, we carried out immunocytochemistry and western blot investigations of CDK4, p-pRB, pRB, and E2F1 in the ADC-NPCs (Fig. 3A and 3K). The immunocytochemical staining results revealed significant changes in the subcellular localization and fluorescence intensity of these cell cycle regulatory proteins (Fig. 3B~3I). We used PCC and MOC to analyze the subcellular localization values and fluorescence intensities of CDK4, pRB, p-pRB, and E2F1 in the ADC-NPCs using the JACoP plugin in ImageJ (Fig. 3B~3E for PCC and Fig. 3F~3I for MOC). CDK4 was mainly expressed in the nuclear fractions of both the wt-NPCs and ADC-NPCs under normal conditions (r=0.51 and 0.49, respectively) and OGD conditions (r=0.4 and 0.38, respectively); the Pearson's r-values indicate that CDK4 and DAPI were closely colocalized (Fig. 3B). The Pearson's r-values for p-pRB expression in the ADC-NPCs under normal and OGD conditions were 0.2 and 0.14 respectively, which indicates weak colocalization with DAPI in the ADC-NPCs. This result indicates that the subcellular localization of p-pRB shifted from the nucleus to the cytoplasm in the ADC-NPCs under normal and OGD conditions (Fig. 3C). However, there were no significant changes in the r-values for the expression of pRB (Fig. 3D), and the subcellular localization of E2F1 had a similar pattern to p-pRB (Fig. 3E; Pearson's r=0.2 and 0.14, respectively). The fluorescence intensity (analyzed by MOC) also revealed that the expression levels of CDK4 (Fig. 3F), p-pRB (Fig. 3G), pRB (Fig. 3H), and E2F1 (Fig. 3I) proteins were significantly increased in the ADC-NPCs following ischemic stress. We carried out western blot analysis of the cytoplasmic and nuclear fractions of the wt-NPCs and ADC-NPCs to evaluate the cytoplasmic and nuclear expression levels of CDK4, p-pRB, pRB, and E2F1 (Fig. 3K). The expression of nuclear CDK4 in the ADC-NPCs was similar to that in the wt-NPCs under both conditions, but the cytoplasmic expression of CDK4 was slightly higher in the ADC-NPCs under normal conditions than under OGD conditions (Fig. 3L). The expression of p-pRB (Ser 807/811) was significantly higher in the cytoplasmic fraction of the ADC-NPCs than in the nuclear fraction under both normal and OGD conditions (Fig. 3M). pRB protein expression increased significantly in the cytoplasmic fractions of both the wt-NPCs and ADC-NPCs under normal conditions, but they were significantly reduced under OGD conditions (Fig. 3N). E2F1 expression was significantly reduced in the cytoplasmic fraction of the ADC-NPCs under both normal and OGD conditions (Fig. 3O). These results suggest that the overexpression of ADC genes in NPCs regulates cell cycle arrest by promoting CDK4-induced pRB phosphorylation and inducing the nucleocytoplasmic shuttling of p-pRB and E2F1 following ischemic stress.
The overexpression of ADC genes in the NPCs promotes cell cycle exit and neural differentiation, but it is not clear which factors regulate such changes and modifications. Accumulating evidence suggests that the increasing level of intracellular Ca2+ plays an essential role in the cell cycle, including cell proliferation, differentiation, and apoptosis [33]. In the present study, we hypothesized that overexpressed ADC genes elevate the intracellular calcium concentration [Ca2+i] in ADC-NPCs following ischemic stress. To investigate the changes in [Ca2+i] levels, we stained the cells with Fluo-4AM (4 µM) and used time-lapse fluorescence imaging (Fig. 4A and 4B). The [Ca2+i] levels increased significantly in the ADC-NPCs in both the resting state (without treatment or treatment with the negative control (dimethyl sulfoxide): 117.37±7.38 nM; Fig. 4A) and excitation state (ionomycin treatment: 288.58±26.96 nM; Fig. 4B) compared to [Ca2+i] levels in the wt-NPCs following OGD injury (resting: 21.19±0.22 nM and excitation: 28.97±1.4 nM). To determine whether these increases in [Ca2+i] levels were caused by the overexpression of ADC genes, we used small interfering human ADC (siADC, 10 nM; Bioneer, Republic of Korea) to interfere with the expression of the ADC genes. We transfected siADC into the ADC-NPCs over 3 days, and labeled the cells with Fluo-4AM (4 µM) to determine the intracellular [Ca2+i] levels (Fig. 4C and 4D). The excessive [Ca2+i] levels observed in the resting and excitation states (136.6±0.27 nM and 505.07±22.49 nM, respectively) were significantly reduced by si-ADC transfection (86.56±0.19 nM and 311.55±18.19 nM, respectively). These results suggest that the overexpression of ADC genes can elevate and regulate intracellular [Ca2+i] levels in NPCs under pathological conditions. However, it is well known that excessive intracellular [Ca2+i] is a major cause of cellular stress, and induces mitochondria-dependent apoptosis [7]. Our previous reports [14,16,20] and the present study have shown that cell death can be attenuated in ADC-NPCs following ischemic insults (Fig. 1C and 1D). Following these results, we investigated the mitochondrial cellular metabolism of the ADC-NPCs. We assessed the biological profile of the OCR (Fig. 4E) as a mitochondrial function in the ADC-NPCs using an XF24 extracellular flux analyzer under both normal and pathological conditions (n=7 replicates from two independent experiments). Interestingly, we found that the basal respiration level of the OCR increased significantly in the ADC-NPCs under normal conditions (695.56±23.33 pMoles/min/µg) compared to the basal respiration levels of the OCR in the wt-NPCs under normal conditions and in the ADC-NPCs under OGD conditions (119.99±37.79 and 434.03±84.78 pMoles/min/µg, respectively) (Fig. 4F). Assessment of the area under the curve of the oxygen consumption rate (OCR-AUC) revealed that the mitochondrial ability of the ADC-NPCs at basal respiration (Fig. 4G), the ATP-linked respiration (Fig. 4H), and the maximal respiration (Fig. 4I) were significantly elevated under normal conditions (7840.23±1817.27, 4029.26±1252.83, and 15053.96±4057.87 pMoles/min, respectively) and under OGD conditions (3055.39±583.34, 1266.85±306.65, and 3928.29±778.31 pMoles/min, respectively). These data suggest that the overexpression of ADC significantly increases the mitochondrial membrane potential (ΔΨm) following ischemic stress (Fig. 4F~4I). Collectively, these data suggest that the overexpression of ADC genes plays a pivotal role in elevating mitochondrial membrane potential and respiration, and increasing intracellular calcium levels in NPCs following ischemic stress.
A growing body of evidence suggests that many signaling proteins are related to NPC differentiation, and are also related to cellular [Ca2+i] and intracellular ionic changes. CREB, ERK1/2, and JAK-STAT signaling are reportedly related to NPC differentiation and calcium homeostasis in cells [34]. Changes to the cellular [Ca2+i] level may facilitate the importation of phosphorylated STAT into the nucleus to induce maximal transcriptional activation, which is also accompanied by the phosphorylation of ERK1/2 (Thr202/Tyr204), and CREB (Ser133) in NPCs [35,36,37,38]. In the present study, we investigated changes in the phosphorylated protein levels and the cellular localization of pERK1/2, pCREB, and pSTAT1 in the ADC-NPCs. The immunocytochemical staining results showed that cytoplasmic pERK1/2, and nuclear pCREB and pSTAT1 proteins were highly expressed in the ADC-NPCs under both normal and ischemic conditions (Fig. 5A). The cellular localizations of pERK1/2 (Fig. 5B), pCREB (Fig. 5C), and pSTAT1 (Fig. 5D) (analyzed by PCC) were determined from the immunocytochemical staining of the wt-NPCs and ADC-NPCs under both normal and ischemic conditions. Interestingly, the Pearson's r-value for pERK1/2 expression was zero (r=0), which implies there was no correlation with DAPI in the ADC-NPCs following OGD injury. This result suggests that pERK1/2 expression was significant in the cytoplasm of the ADC-NPCs under OGD conditions. Moreover, the Pearson's r-value for pSTAT1 expression was r=0.8 in the ADC-NPCs following OGD injury, which means that pSTAT1 was highly colocalized with DAPI, suggesting that there is a significant accumulation of pSTAT1 in the nuclei of the ADC-NPCs during ischemic stress (Fig. 5F and 5G). The fluorescence intensity (analyzed by MOC) indicated that the expression levels of pERK1/2 (Fig. 5E), pCREB (Fig. 5F), and pSTAT1 (Fig. 5G) proteins increased significantly in the ADC-NPCs under both normal and ischemic conditions. The results of western blot analysis corroborated those from the immunocytochemical staining of the wt-NPCs and ADC-NPCs (Fig. 5H~5N). The quantification of the western blot data for the ADC-NPCs showed significantly higher expression levels of cytoplasmic pERK1/2, nuclear pCREB, and pSTAT1 under ischemic conditions. In summary, the results of the
To determine the therapeutic potential of ADC-NPCs in the CNS, we injected ADC-NPCs into the brain of an adult rat following cerebral ischemic injury, and determined the infarct volume, neurological functional recovery, neuronal differentiation, and synapse formation. The ADC-NPCs were transplanted into the rat brain 7 days after MCAO, and had significantly reduced the brain infarct volume by Days 14 and 28 after MCAO (Fig. 6A). Motor function had increased significantly by Day 14 after MCAO in the ADC-NPC-transplanted groups compared to the experimental control (EC) and wt-NPCs groups, and continued to increase until Day 28 after MCAO (Fig. 6B). The immunohistochemical staining of the ADC-NPC-transplanted rats revealed marked increases in the expression levels of the mature neuron marker MAP2+ both in and beyond the peri-infarct areas by Day 14 after MCAO, compared to the EC- and normal NPC-transplanted rats (Fig. 6C). The levels of the synaptic molecules synaptophysin and PSD95, levels of the neuronal marker GAP43, and the number of neurofilaments increased in the ADC-NPC-transplanted groups (Fig. 6D~6H). The colocalization of synaptophysin and PSD95 in the MAP2-positive cells and the number of synaptophysin puncta also increased significantly in the ADC-NPC-transplanted groups (Fig. 6I and 6J). Collectively, the results demonstrate that the transplanted ADC-NPCs can attenuate infarct volume, promote neural differentiation, and enhance motor function recovery, and the synapse formation and transplantation of ADC-NPCs suggests their potential as the basis of a stem cell therapy for cerebral ischemic injury.
DISCUSSION
Cell replacement therapy using NPCs could become a popular option for treating ischemic brain injury. However, transplanted NPCs often encounter harsh environmental conditions in the ischemic brain, and it is difficult to control their fate. In the present study, we identified the cellular characteristics of NPCs overexpressing ADC genes (ADC-NPCs) during ischemic stress. Cell death was significantly reduced in the ADC-NPCs, and proliferation potential was suppressed by intracellular Ca2+-induced CDK4 activation and nucleocytoplasmic shuttling of p-pRB and E2F1 in the ADC-NPCs following OGD injury. Moreover, the ADC-NPCs were attenuated by excessive cytosolic Ca2+-induced mitochondrial dysfunction and cell death following ischemic stress. Consequently, molecular cascades in the ADC-NPCs promoted neuronal differentiation by enhancing pSTAT1 (ser727) expression via Ca2+-related ERK1/2 and CREB phosphorylation. The transplantation of these ADC-NPCs promoted synapse formation and motor function recovery following cerebral ischemia. Therefore, our study provides the basis for a novel cell replacement therapy for ischemic stroke using ADC-NPCs that could attenuate excessive calcium tolerance, mitochondrial dysfunction, and behavior retardation.
Previously it was reported that the arginine decarboxylase (ADC) is a part of the acid-resistance (AR) system in the several organisms such as
The short-term OGD exposure induces the neural differentiation of NPCs while maintaining cell survival [46,47]. The expression of neuronal differentiation marker DCX and oligodendroglial marker Olig2 were increased, but the expression of astrocyte marker GFAP was reduced in the ADC-NPCs following ischemic stress. One of the candidate neural differentiation and glial inhibitory mechanism is BMP signaling. BMPs are known to regulate proliferation or differentiation of neurons, oligodendrocytes, and astrocytes during CNS development [48]. Previous our study suggests that ADC expression and agmatine treatment reduced collagen scar formation and functional outcome may due to increased tissue preservation within the injury site in SCI mice [19,49]. In particular, BMP4 enhanced the astrocytic lineage during CNS development and contributed to the formation of glial scar in injured spinal cord [50,51]. Following these previous reports suggesting that the reduction of differentiation to astrocytes in ADC-NPCs is expected to lead to a decrease in glial scar formation following cerebral ischemic injury, and increased differentiation into neurons and oligodendrocytes in ADC-NPCs will also lead to axonal regeneration and functional recovery. It has also shown that exogenously administered agmatine to SVZ-NSC cells increases neuronal differentiation but decreases astrogenesis [52]. It has reported that this neurogesis effect of agmatine seem to be related to the extracellular signal-regulated kinase (ERK1/2) activation, and the anti-astrogenesis effect is related to inhibition of bone morphogenetic proteins (BMPs) [52]. In addition, Marchal et al. [53] and Leclerc [54,55,56] et al. provide with intriguing clues as to the relationship among BMP signal blocking, FGF signaling and calcium signaling in neuroectodermal differentiation. The BMP signal blocking is responsible to the neuroectodermal differentiation by calcium signaling [57]. Although this study did not show a direct correlation between increased calcium concentration and BMP signaling in ADC-NPCs, based on the results of this study and previous research reports, we might suggest that neural and glial differentiation in ADC-NPCs is regulated through BMP and calcium signaling.
Calcium acts as a pleiotropic second messenger that triggers numerous physiological processes such as cell proliferation, differentiation, and cell death [58,59]. CDKs, pRB, and E2F1 are well-known transcriptional regulators that determine cell cycle progression during the G1-S phase transition [60]. Hypophosphorylated pRB recognizes E2F-DP complexes, which act as tumor suppressors by inhibiting the G1-S phase checkpoint and controlling differentiation and apoptosis [61,62]. Mammalian cells contain various CDKs and cyclins, and oscillations in the expression and activity of these cell cycle molecules are regulated during cell division [63,64]. During cell cycle progression, Ca2+ assembles and activates the cyclinD-CDK4 and cyclinE-CDK2 complexes. Later in the G1 phase, Ca2+ ensures the phosphorylation of retinoblastoma protein (p-pRB) in the nucleus and entry into the S phase. Moreover, calmodulin-induced CDK4 activation regulates the nuclear export of phosphorylated pRB in mouse embryo fibroblasts [65], and E2F1 induces nucleocytoplasmic shuttling in high Ca2+ conditions in mouse keratinocytes. Endogenous E2F1 is substantially downregulated during differentiation [66,67]. The results of the present study suggest that overexpressed ADC genes in ADC-NPCs promote higher levels of p-pRB and suppress E2F1 expression in the cytoplasmic fraction, which might terminate the cell cycle and initiate cellular differentiation. The reason for the higher p-pRB expression in the cytoplasm of the ADC-NPCs requires further investigation. One of the possible explanations for this phenomenon is the regulation of cytosolic calcium ions (Ca2+), which reportedly control the cell cycle via a Ca2+/calmodulin (CaM) complex that regulates the expression of cell cycle molecules such as CDKs [59,63,68,69]. Ca2+ is a ubiquitous secondary messenger that is involved in the regulation of many cellular activities. Importantly, both cytosolic and nuclear Ca2+ levels play essential roles in cell cycle progression, cell proliferation, differentiation, and apoptosis [33]. Moreover, one of the many roles of Ca2+ is as a key regulator of mitochondrial function that controls ATP synthesis. However, the dysregulation of Ca2+ homeostasis leads to the enhanced generation of ROS, triggering the opening of mitochondrial permeability transition pores (MPTPs) and cytochrome c release leading to apoptosis [70]. According to previous studies, the metabolite of ADC, agmatine, has a scavenging effect on toxic ROS by protecting against a drop in energy capacity, Ca2+-induced mitochondrial swelling, Ca2+-dependent induction of mitochondrial permeability, and mitochondrial membrane potential (ΔΨm) collapse in rat brain and rat kidney mitochondria [12,13]. Our results indicate that the level of cellular Ca2+ was upregulated in the ADC-NPCs during OGD. This increase in cellular Ca2+ was suppressed by ADC-specific siRNA transfection, and it was evident that the increase in the Ca2+ levels in the ADC-NPCs was caused by the overexpression of ADC genes. However, the accumulation of intracellular calcium in the cytosol of the ADC-NPCs was not accompanied by mitochondrial dysfunction owing to ROS scavenging in the presence of endogenous agmatine production. In the present study, the mitochondrial OCR in the wt-NPCs was significantly reduced the mitochondrial respiration rates, ATP synthesis and accompanied with cell death. But, the mitochondrial OCR in the ADC-NPCs was significantly higher during the basal mitochondrial respiration, ATP-linked respiration, and maximal respiration under both normal and OGD conditions. Therefore, we suggest that overexpressed ADC genes in ADC-NPCs might play a vital role in strengthening intracellular calcium resistance and preventing mitochondrial membrane potential collapse following ischemic injury.
Phosphorylation of the transcriptional factor STAT1/3 plays an important role in the CNS during neuroglial differentiation [34,71,72]. Previous reports suggest that STAT1 can become phosphorylated in response to cellular stress involving elevated ERK and [Ca2+i] levels [73,74,75]. Furthermore, Takagi et al. reported that STAT1 phosphorylation at Tyr701 and Ser727 (pSTAT1), nuclear translocation, and DNA binding can be promoted within hours of cerebral ischemia [76]. Phosphorylated ERK1/2 is involved in regulating STAT1/3 function [36]. Phosphorylated CREB is usually absent in adult stem cells, and is transiently upregulated in NPCs in the differentiation state [77]. CREB phosphorylated at Ser133 (pCREB) is involved in regulating various signaling pathways triggered by a variety of factors that cause cellular stress, including H2O2 exposure, UV light, inflammatory cytokines, and growth factors [78,79]. The loss of CREB signaling results in cell death and the loss of neuronal gene expression [80]. These multiple roles of CREB may be controlled by the phosphorylation of Ca2+/calmodulin-dependent protein kinases (CaMKs). Our study revealed a significant increase in the level of pSTAT1 protein following ERK1/2 and CREB phosphorylation in the ADC-NPCs after ischemic injury. pCREB phosphorylated at Ser133 promotes transcriptional activity and interacts with NGN1, forming the CREB-binding protein (CBP)/P300-pSTAT1/3 complex. pSTAT1 could increase the transcriptional activities of neuronal-specific genes (e.g., NGN, DCX, Olig2), and the CBP/P300 component of this complex is recognized by the bHLH DNA-binding motif of the proneural gene of an E-box site via a bZIP transcriptional factor, binding to neurogenin1 and neurogenin2 [81,82]. Besides, the treatment of ischemic stroke by cell replacement therapy involves checking tissue and cellular damage, neuroglial replacement to restructure neuronal connectivity and conduction, and the provision of a favorable tropic environment around the ischemic area [83,84]. In our previous study, it has reported that ADC-MSCs promote locomotor function after SCI by increasing BDNF level, which has been associated with neuronal survival and replacement of damaged neurons in SCI and exerts a paracrine effect. This finding may elucidate the paracrine effects of BDNF as key roles in the therapeutic effects on the neuronal regeneration, oligodendrogenesis, inhibition of glial scar formation, improved motor function recovery in ADC-transfected cells following CNS injury [19]. Based on the previous studies, even we did not confirm the BDNF level in the ADC-NPCs transplantation after cerebral ischemia, it is possible that the ADC-NPCs has therapeutic effects via BDNF expression, and activates neural differentiation through the BDNF-induced phosphorylation of pCREB/pSTAT1 after CNS injury.
The transplantation of ADC-NPCs reduced the infarct volume compared to the transplantation of wt-NPCs following transient cerebral ischemic injury. Motor function was also markedly restored in the rats that had received ADC-NPC transplantation compared to those that had not. Moreover, there was greater neural differentiation and neuronal fiber formation, and the number of synaptophysin puncta increased significantly in the peri-infarct region of the ADC-NPC groups following cerebral ischemia. In the present study, the protein expression of synaptophysin, GAP43, PSD95, and neurofilament were significantly increased in the ipsilateral brain transplanted ADC-NPCs at 14 days after MCAO. The GAP43 is nervous system-specific protein, is highly expressed neuronal growth cones during neurogenesis [85]. The GAP43 expression was highly correlated with synaptophysin, PSD95, and neurofilament in the transplanted ADC-NPCs group. However, we have some limitation about this study, which is identified the origin of the cells in the ipsilateral brain. We were used BrdU incorporation for
In conclusion, we have reported the therapeutic potential of ADC-NPCs, which promote cell cycle exit and neural differentiation by elevating [Ca2+i]-induced CDK4 activation, nucleocytoplasmic shuttling of pRB-E2F1, and nuclear translocation of pSTAT1 following ischemic stress. Moreover, the excessive intracellular calcium-induced mitochondrial membrane potential collapse following ischemic stress was prevented in the ADC-NPCs. Transplanted ADC-NPC promoted neurological functional recovery following ischemic stroke. This may have been owing to neural differentiation and synapse formation in a favorable environment. The present study indicates that the overexpression of ADC genes in ADC-NPCs could form the basis of a promising cell replacement therapy.
Figures

{kind=link}

{kind=link}

{kind=link}

{kind=link}

{kind=link}

{kind=link}
References
- Bacigaluppi M, Russo GL, Peruzzotti-Jametti L, Rossi S, Sandrone S, Butti E, De Ceglia R, Bergamaschi A, Motta C, Gallizioli M, Studer V, Colombo E, Farina C, Comi G, Politi LS, Muzio L, Villani C, Invernizzi RW, Hermann DM, Centonze D, Martino G. Neural stem cell transplantation induces stroke recovery by upregulating glutamate transporter GLT-1 in astrocytes. J Neurosci 2016;36:10529-10544.

- Blurton-Jones M, Kitazawa M, Martinez-Coria H, Castello NA, Müller FJ, Loring JF, Yamasaki TR, Poon WW, Green KN, LaFerla FM. Neural stem cells improve cognition via BDNF in a transgenic model of Alzheimer disease. Proc Natl Acad Sci U S A 2009;106:13594-13599.
- Braccioli L, Heijnen CJ, Coffer PJ, Nijboer CH. Delayed administration of neural stem cells after hypoxia-ischemia reduces sensorimotor deficits, cerebral lesion size, and neuroinflammation in neonatal mice. Pediatr Res 2017;81:127-135.
- Daadi MM, Davis AS, Arac A, Li Z, Maag AL, Bhatnagar R, Jiang K, Sun G, Wu JC, Steinberg GK. Human neural stem cell grafts modify microglial response and enhance axonal sprouting in neonatal hypoxic-ischemic brain injury. Stroke 2010;41:516-523.
- Martino G, Pluchino S. The therapeutic potential of neural stem cells. Nat Rev Neurosci 2006;7:395-406.
- Park KI, Teng YD, Snyder EY. The injured brain interacts reciprocally with neural stem cells supported by scaffolds to reconstitute lost tissue. Nat Biotechnol 2002;20:1111-1117.
- Doyle KP, Simon RP, Stenzel-Poore MP. Mechanisms of ischemic brain damage. Neuropharmacology 2008;55:310-318.
- Thompson BJ, Ronaldson PT. Drug delivery to the ischemic brain. Adv Pharmacol 2014;71:165-202.
- Szydlowska K, Tymianski M. Calcium, ischemia and excitotoxicity. Cell Calcium 2010;47:122-129.
- Kumar BK. In: Lee JE. Stem cells and cancer stem cells. Hayat MA, ed. Dordrecht: Springer, 2012; 2012. p. 195-202.
- Piletz JE, Aricioglu F, Cheng JT, Fairbanks CA, Gilad VH, Haenisch B, Halaris A, Hong S, Lee JE, Li J, Liu P, Molderings GJ, Rodrigues AL, Satriano J, Seong GJ, Wilcox G, Wu N, Gilad GM. Agmatine: clinical applications after 100 years in translation. Drug Discov Today 2013;18:880-893.
- Arndt MA, Battaglia V, Parisi E, Lortie MJ, Isome M, Baskerville C, Pizzo DP, Ientile R, Colombatto S, Toninello A, Satriano J. The arginine metabolite agmatine protects mitochondrial function and confers resistance to cellular apoptosis. Am J Physiol Cell Physiol 2009;296:C1411-C1419.
- Battaglia V, Grancara S, Satriano J, Saccoccio S, Agostinelli E, Toninello A. Agmatine prevents the Ca(2+)-dependent induction of permeability transition in rat brain mitochondria. Amino Acids 2010;38:431-437.
- Bokara KK, Kwon KH, Nho Y, Lee WT, Park KA, Lee JE. Retroviral expression of arginine decarboxylase attenuates oxidative burden in mouse cortical neural stem cells. Stem Cells Dev 2011;20:527-537.
- Bokara KK, Kim JH, Kim JY, Lee JE. Transfection of arginine decarboxylase gene increases the neuronal differentiation of neural progenitor cells. Stem Cell Res (Amst) 2016;17:256-265.
- Hong S, Son MR, Yun K, Lee WT, Park KA, Lee JE. Retroviral expression of human arginine decarboxylase reduces oxidative stress injury in mouse cortical astrocytes. BMC Neurosci 2014;15:99.
- Kim JH, Yenari MA, Giffard RG, Cho SW, Park KA, Lee JE. Agmatine reduces infarct area in a mouse model of transient focal cerebral ischemia and protects cultured neurons from ischemia-like injury. Exp Neurol 2004;189:122-130.
- Kim JH, Lee YW, Park KA, Lee WT, Lee JE. Agmatine attenuates brain edema through reducing the expression of aquaporin-1 after cerebral ischemia. J Cereb Blood Flow Metab 2010;30:943-949.
- Park YM, Han SH, Seo SK, Park KA, Lee WT, Lee JE. Restorative benefits of transplanting human mesenchymal stromal cells overexpressing arginine decarboxylase genes after spinal cord injury. Cytotherapy 2015;17:25-37.
- Moon SU, Kwon KH, Kim JH, Bokara KK, Park KA, Lee WT, Lee JE. Recombinant hexahistidine arginine decarboxylase (hisADC) induced endogenous agmatine synthesis during stress. Mol Cell Biochem 2010;345:53-60.
- Lee WT, Hong S, Yoon SH, Kim JH, Park KA, Seong GJ, Lee JE. Neuroprotective effects of agmatine on oxygen-glucose deprived primary-cultured astrocytes and nuclear translocation of nuclear factor-kappa B. Brain Res 2009;1281:64-70.
- Karuppagounder SS, Basso M, Sleiman SF, Ma TC, Speer RE, Smirnova NA, Gazaryan IG, Ratan RR. In vitro ischemia suppresses hypoxic induction of hypoxia-inducible factor-1α by inhibition of synthesis and not enhanced degradation. J Neurosci Res 2013;91:1066-1075.
- Iyo AH, Zhu MY, Ordway GA, Regunathan S. Expression of arginine decarboxylase in brain regions and neuronal cells. J Neurochem 2006;96:1042-1050.
- Jung H, Toth PT, White FA, Miller RJ. Monocyte chemoattractant protein-1 functions as a neuromodulator in dorsal root ganglia neurons. J Neurochem 2008;104:254-263.
- Helmchen F. Calibration protocols for fluorescent calcium indicators. Cold Spring Harb Protoc 2011;2011:980-984.
- Bolte S, Cordelières FP. A guided tour into subcellular colocalization analysis in light microscopy. J Microsc 2006;224:213-232.
- Manders EM, Stap J, Brakenhoff GJ, van Driel R, Aten JA. Dynamics of three-dimensional replication patterns during the S-phase, analysed by double labelling of DNA and confocal microscopy. J Cell Sci 1992;103:857-862.
- Manders EM, Verbeek FJ, Aten JA. Measurement of co-localization of objects in dual-colour confocal images. J Microsc 1993;169:375-382.
- Caster AH, Kahn RA. Computational method for calculating fluorescence intensities within three-dimensional structures in cells. Cell Logist 2012;2:176-188.
- Budirahardja Y, Gönczy P. Coupling the cell cycle to development. Development 2009;136:2861-2872.
- Malumbres M, Barbacid M. Cell cycle, CDKs and cancer: a changing paradigm. Nat Rev Cancer 2009;9:153-166.
- van den Heuvel S, Dyson NJ. Conserved functions of the pRB and E2F families. Nat Rev Mol Cell Biol 2008;9:713-724.
- Berridge MJ, Bootman MD, Roderick HL. Calcium signalling: dynamics, homeostasis and remodelling. Nat Rev Mol Cell Biol 2003;4:517-529.
- Deverman BE, Patterson PH. Cytokines and CNS development. Neuron 2009;64:61-78.
- Toth AB, Shum AK, Prakriya M. Regulation of neurogenesis by calcium signaling. Cell Calcium 2016;59:124-134.
- Li Z, Theus MH, Wei L. Role of ERK 1/2 signaling in neuronal differentiation of cultured embryonic stem cells. Dev Growth Differ 2006;48:513-523.
- Wen Z, Zhong Z, Darnell JE. Maximal activation of transcription by Stat1 and Stat3 requires both tyrosine and serine phosphorylation. Cell 1995;82:241-250.
- Kovarik P, Stoiber D, Eyers PA, Menghini R, Neininger A, Gaestel M, Cohen P, Decker T. Stress-induced phosphorylation of STAT1 at Ser727 requires p38 mitogen-activated protein kinase whereas IFN-gamma uses a different signaling pathway. Proc Natl Acad Sci U S A 1999;96:13956-13961.
- Auger EA, Redding KE, Plumb T, Childs LC, Meng SY, Bennett GN. Construction of lac fusions to the inducible arginine- and lysine decarboxylase genes of Escherichia coli K12. Mol Microbiol 1989;3:609-620.
- Castanie-Cornet MP, Penfound TA, Smith D, Elliott JF, Foster JW. Control of acid resistance in Escherichia coli. J Bacteriol 1999;181:3525-3535.
- Kanjee U, Houry WA. Mechanisms of acid resistance in Escherichia coli. Annu Rev Microbiol 2013;67:65-81.
- Lin J, Lee IS, Frey J, Slonczewski JL, Foster JW. Comparative analysis of extreme acid survival in Salmonella typhimurium, Shigella flexneri, and Escherichia coli. J Bacteriol 1995;177:4097-4104.
- Shi X, Waasdorp BC, Bennett GN. Modulation of acid-induced amino acid decarboxylase gene expression by hns in Escherichia coli. J Bacteriol 1993;175:1182-1186.
- Seo SK, Yang W, Park YM, Lee WT, Park KA, Lee JE. Overexpression of human arginine decarboxylase rescues human mesenchymal stem cells against H2O2 toxicity through cell survival protein activation. J Korean Med Sci 2013;28:366-373.
- Kim JH, Kim JY, Jung JY, Lee YW, Lee WT, Huh SK, Lee JE. Endogenous agmatine induced by ischemic preconditioning regulates ischemic tolerance following cerebral ischemia. Exp Neurobiol 2017;26:380-389.
- Wang Q, Yang L, Wang Y. Enhanced differentiation of neural stem cells to neurons and promotion of neurite outgrowth by oxygen-glucose deprivation. Int J Dev Neurosci 2015;43:50-57.
- Shen C, Cheng W, Yu P, Wang L, Zhou L, Zeng L, Yang Q. Resveratrol pretreatment attenuates injury and promotes proliferation of neural stem cells following oxygen-glucose deprivation/reoxygenation by upregulating the expression of Nrf2, HO-1 and NQO1 in vitro. Mol Med Rep 2016;14:3646-3654.
- Ille F, Atanasoski S, Falk S, Ittner LM, Märki D, Büchmann-Møller S, Wurdak H, Suter U, Taketo MM, Sommer L. Wnt/BMP signal integration regulates the balance between proliferation and differentiation of neuroepithelial cells in the dorsal spinal cord. Dev Biol 2007;304:394-408.
- Park YM, Lee WT, Bokara KK, Seo SK, Park SH, Kim JH, Yenari MA, Park KA, Lee JE. The multifaceted effects of agmatine on functional recovery after spinal cord injury through Modulations of BMP-2/4/7 expressions in neurons and glial cells. PLoS One 2013;8:e53911.
- Gomes WA, Mehler MF, Kessler JA. Transgenic overexpression of BMP4 increases astroglial and decreases oligodendroglial lineage commitment. Dev Biol 2003;255:164-177.
- White RE, Jakeman LB. Don't fence me in: harnessing the beneficial roles of astrocytes for spinal cord repair. Restor Neurol Neurosci 2008;26:197-214.
- Song HW, Kumar BK, Kim SH, Jeon YH, Lee YA, Lee WT, Park KA, Lee JE. Agmatine enhances neurogenesis by increasing ERK1/2 expression, and suppresses astrogenesis by decreasing BMP 2,4 and SMAD 1,5,8 expression in subventricular zone neural stem cells. Life Sci 2011;89:439-449.
- Marchal L, Luxardi G, Thomé V, Kodjabachian L. BMP inhibition initiates neural induction via FGF signaling and Zic genes. Proc Natl Acad Sci U S A 2009;106:17437-17442.
- Batut J, Vandel L, Leclerc C, Daguzan C, Moreau M, Néant I. The Ca2+-induced methyltransferase xPRMT1b controls neural fate in amphibian embryo. Proc Natl Acad Sci U S A 2005;102:15128-15133.
- Leclerc C, Webb SE, Daguzan C, Moreau M, Miller AL. Imaging patterns of calcium transients during neural induction in Xenopus laevis embryos. J Cell Sci 2000;113:3519-3529.
- Leclerc C, Lee M, Webb SE, Moreau M, Miller AL. Calcium transients triggered by planar signals induce the expression of ZIC3 gene during neural induction in Xenopus. Dev Biol 2003;261:381-390.
- Aruga J, Mikoshiba K. Role of BMP, FGF, calcium signaling, and Zic proteins in vertebrate neuroectodermal differentiation. Neurochem Res 2011;36:1286-1292.
- Berridge MJ, Lipp P, Bootman MD. The versatility and universality of calcium signalling. Nat Rev Mol Cell Biol 2000;1:11-21.
- Humeau J, Bravo-San Pedro JM, Vitale I, Nuñez L, Villalobos C, Kroemer G, Senovilla L. Calcium signaling and cell cycle: progression or death. Cell Calcium 2018;70:3-15.
- Bertoli C, Skotheim JM, de Bruin RA. Control of cell cycle transcription during G1 and S phases. Nat Rev Mol Cell Biol 2013;14:518-528.
- Ikeda MA, Jakoi L, Nevins JR. A unique role for the Rb protein in controlling E2F accumulation during cell growth and differentiation. Proc Natl Acad Sci U S A 1996;93:3215-3220.
- Shirodkar S, Ewen M, DeCaprio JA, Morgan J, Livingston DM, Chittenden T. The transcription factor E2F interacts with the retinoblastoma product and a p107-cyclin A complex in a cell cycle-regulated manner. Cell 1992;68:157-166.
- Morgan DO. Principles of CDK regulation. Nature 1995;374:131-134.
- Lim S, Kaldis P. Cdks, cyclins and CKIs: roles beyond cell cycle regulation. Development 2013;140:3079-3093.
- Jiao W, Datta J, Lin HM, Dundr M, Rane SG. Nucleocytoplasmic shuttling of the retinoblastoma tumor suppressor protein via Cdk phosphorylation-dependent nuclear export. J Biol Chem 2006;281:38098-38108.
- Ivanova IA, Dagnino L. Activation of p38- and CRM1-dependent nuclear export promotes E2F1 degradation during keratinocyte differentiation. Oncogene 2007;26:1147-1154.
- Ivanova IA, Vespa A, Dagnino L. A novel mechanism of E2F1 regulation via nucleocytoplasmic shuttling: determinants of nuclear import and export. Cell Cycle 2007;6:2186-2195.
- Sherr CJ. Cancer cell cycles. Science 1996;274:1672-1677.
- Hunter T, Pines J. Cyclins and cancer. II: cyclin D and CDK inhibitors come of age. Cell 1994;79:573-582.
- Brookes PS, Yoon Y, Robotham JL, Anders MW, Sheu SS. Calcium, ATP, and ROS: a mitochondrial love-hate triangle. Am J Physiol Cell Physiol 2004;287:C817-C833.
- Hofmann HD, Kirsch M. JAK2-STAT3 signaling: a novel function and a novel mechanism. JAKSTAT 2012;1:191-193.
- Kanski R, van Strien ME, van Tijn P, Hol EM. A star is born: new insights into the mechanism of astrogenesis. Cell Mol Life Sci 2014;71:433-447.
- Goh KC, Haque SJ, Williams BR. p38 MAP kinase is required for STAT1 serine phosphorylation and transcriptional activation induced by interferons. EMBO J 1999;18:5601-5608.
- Nair JS, DaFonseca CJ, Tjernberg A, Sun W, Darnell JE, Chait BT, Zhang JJ. Requirement of Ca2+ and CaMKII for Stat1 Ser-727 phosphorylation in response to IFNgamma. Proc Natl Acad Sci U S A 2002;99:5971-5976.
- Uddin S, Sassano A, Deb DK, Verma A, Majchrzak B, Rahman A, Malik AB, Fish EN, Platanias LC. Protein kinase C-delta (PKC-delta) is activated by type I interferons and mediates phosphorylation of Stat1 on serine 727. J Biol Chem 2002;277:14408-14416.
- Takagi Y, Harada J, Chiarugi A, Moskowitz MA. STAT1 is activated in neurons after ischemia and contributes to ischemic brain injury. J Cereb Blood Flow Metab 2002;22:1311-1318.
- Merz K, Herold S, Lie DC. CREB in adult neurogenesis--master and partner in the development of adult-born neurons?. Eur J Neurosci 2011;33:1078-1086.
- Xing J, Kornhauser JM, Xia Z, Thiele EA, Greenberg ME. Nerve growth factor activates extracellular signal-regulated kinase and p38 mitogen-activated protein kinase pathways to stimulate CREB serine 133 phosphorylation. Mol Cell Biol 1998;18:1946-1955.
- Barlow CA, Kitiphongspattana K, Siddiqui N, Roe MW, Mossman BT, Lounsbury KM. Protein kinase A-mediated CREB phosphorylation is an oxidant-induced survival pathway in alveolar type II cells. Apoptosis 2008;13:681-692.
- Herold S, Jagasia R, Merz K, Wassmer K, Lie DC. CREB signalling regulates early survival, neuronal gene expression and morphological development in adult subventricular zone neurogenesis. Mol Cell Neurosci 2011;46:79-88.
- Ma Z, Chang MJ, Shah RC, Benveniste EN. Interferon-gamma-activated STAT-1alpha suppresses MMP-9 gene transcription by sequestration of the coactivators CBP/p300. J Leukoc Biol 2005;78:515-523.
- Wojciak JM, Martinez-Yamout MA, Dyson HJ, Wright PE. Structural basis for recruitment of CBP/p300 coactivators by STAT1 and STAT2 transactivation domains. EMBO J 2009;28:948-958.
- Burns TC, Verfaillie CM, Low WC. Stem cells for ischemic brain injury: a critical review. J Comp Neurol 2009;515:125-144.
- Locatelli F, Bersano A, Ballabio E, Lanfranconi S, Papadimitriou D, Strazzer S, Bresolin N, Comi GP, Corti S. Stem cell therapy in stroke. Cell Mol Life Sci 2009;66:757-772.
- Aigner L, Caroni P. Depletion of 43-kD growth-associated protein in primary sensory neurons leads to diminished formation and spreading of growth cones. J Cell Biol 1993;123:417-429.
- Sauerzweig S, Baldauf K, Braun H, Reymann KG. Time-dependent segmentation of BrdU-signal leads to late detection problems in studies using BrdU as cell label or proliferation marker. J Neurosci Methods 2009;177:149-159.