Articles

Article Tools
 View Full Text View Full Text |
 Abstract Abstract |
 Article as PDF Article as PDF |
 Print this Article Print this Article |
 Pubmed Pubmed |
 PMC PMC |
 PubReader PubReader |
 Export to Citation Export to Citation |
 Email Alerts Email Alerts |
 Open Access Open Access |
Share this article on :



Stats or Metrics
Article
Original Article
Exp Neurobiol 2019; 28(3): 390-403
Published online June 26, 2019
https://doi.org/10.5607/en.2019.28.3.390
© The Korean Society for Brain and Neural Sciences
Memantine Improves Cognitive Function and Alters Hippocampal and Cortical Proteome in Triple Transgenic Mouse Model of Alzheimer's Disease
Xinhua Zhou1,†, Liang Wang2,†, Wei Xiao3, Zhiyang Su4, Chengyou Zheng4, Zaijun Zhang4, Yuqiang Wang4, Benhong Xu5, Xifei Yang5*, and Maggie Pui Man Hoi1*
1State Key Laboratory of Quality Research in Chinese Medicine and Institute of Chinese Medical Sciences, University of Macau, Macau SAR 999078, China.
2Institute of Biomedical and Pharmaceutical Sciences, Guangdong University of Technology, Guangzhou 510000, China.
3College of Letters & Science, University of Wisconsin-Madison, Madison, WI 53706, USA.
4Institute of New Drug Research and Guangdong Province Key Laboratory of Pharmacodynamic Constituents of Traditional Chinese Medicine, Jinan University College of Pharmacy, Guangzhou 510000, China.
5Key Laboratory of Modern Toxicology of Shenzhen, Shenzhen Center for Disease Control and Prevention, Shenzhen 518000, China.
Correspondence to: *To whom correspondence should be addressed.
Xifei Yang, TEL: 86-0755-2550-8584, FAX: 86-0755-2550-8584, e-mail: xifeiyang@gmail.com
Maggie Pui Man Hoi, TEL: 853-8822-4876, FAX: 853-2884-1358, e-mail: maghoi@um.edu.mo
†These authors contributed equally to this work.
Abstract
Memantine is a non-competitive N-methyl-D-aspartate receptor (NMDAR) antagonist clinically approved for moderate-to-severe Alzheimer's disease (AD) to improve cognitive functions. There is no report about the proteomic alterations induced by memantine in AD mouse model yet. In this study, we investigated the protein profiles in the hippocampus and the cerebral cortex of AD-related transgenic mouse model (3×Tg-AD) treated with memantine. Mice (8-month) were treated with memantine (5 mg/kg/bid) for 4 months followed by behavioral and molecular evaluation. Using step-down passive avoidance (SDA) test, novel object recognition (NOR) test and Morris water maze (MWM) test, it was observed that memantine significantly improved learning and memory retention in 3xTg-AD mice. By using quantitative proteomic analysis, 3301 and 3140 proteins in the hippocampus and the cerebral cortex respectively were identified to be associated with AD abnormalities. In the hippocampus, memantine significantly altered the expression levels of 233 proteins, among which PCNT, ATAXIN2, TNIK, and NOL3 were up-regulated, and FLNA, MARK 2 and BRAF were down-regulated. In the cerebral cortex, memantine significantly altered the expression levels of 342 proteins, among which PCNT, PMPCB, CRK, and MBP were up-regulated, and DNM2, BRAF, TAGLN 2 and FRY1 were down-regulated. Further analysis with bioinformatics showed that memantine modulated biological pathways associated with cytoskeleton and ErbB signaling in the hippocampus, and modulated biological pathways associated with axon guidance, ribosome, cytoskeleton, calcium and MAPK signaling in the cerebral cortex. Our data indicate that memantine induces higher levels of proteomic alterations in the cerebral cortex than in the hippocampus, suggesting memantine affects various brain regions in different manners. Our study provides a novel view on the complexity of protein responses induced by memantine in the brain of AD.
Graphical Abstract
Keywords: Alzheimer's disease (AD), Memantine, Proteomic, MARK2
INTRODUCTION
Alzheimer's disease (AD) is a devastating neurodegenerative disorder with progressive cognitive declines resulting in severe dementia [1]. According to the World Alzheimer Report 2018, more than 50 million people are affected by AD worldwide, and the number is predicted to double every 20 years [2]. The histopathological hallmarks of AD include extracellular accumulation of amyloid beta (Aβ) plaques and intracellular formation of neurofibrillary tangles (NFTs) composed of hyperphosphorylated tau (p-tau) in the hippocampus and the cerebral cortex [3]. Various hypotheses including Aβ pathology, tau pathology, mitochondrial dysfunction, oxidative stress, neuroinflammation and neurotransmitter disturbances have been put forward, however, the pathogenesis of AD remains obscure [4]. Until now there is no disease-modifying therapy for AD, and only five prescription drugs are currently approved by the FDA to treat its symptoms. One of the five drugs is memantine, a non-competitive N-methyl-D-aspartate receptor (NMDAR) antagonist, which is approved for moderate-to-severe AD to improve learning and memory [5].
Memantine binds to NMDAR with moderate affinity [5]. NMDAR is an ionotropic glutamate receptor found in nerve cells activated upon glutamate and glycine binding. Activation of NMDAR results in the opening of a non-selective cation channel with high Ca2+ permeability, which is very important for the regulation of synaptic plasticity and memory function. NMDAR also displays voltage-dependent channel block by external Mg2+ [6]. It is suggested that in AD patients elevated glutamate release from presynaptic neurons induces NMDAR over-activation, causing Ca2+ overload in postsynaptic neurons and resulting in excitotoxicity. It is further followed by desensitization and internalization of NMDA/AMPA, resulting in synaptic depression and dysfunction [7,8]. Various factors and processes have been shown to induce elevated glutamate release, including soluble Aβ oligomers, Aβ plaques, NFTs, mitochondrial dysfunction and oxidative stress [9,10].
It has been shown that memantine could reduce Aβ-mediated glutamate toxicity, tau hyperphosphorylation and amyloid precursor protein (APP) production in human neuroblastoma SK-N-SH cells [11], reduce Aβ1-42 secretion and senile plaque deposition in primary cortical neuronal cells [9,12], and reduce Aβ-mediated ROS injury in primary hippocampal neurons [13]. Memantine has been shown to reduce soluble Aβ1-42 level, Aβ plaque deposition and synaptic loss in APP/PS1 mice [4,14,15], and decrease total tau and hyperphosphorylated tau in 3×Tg-AD mice [3]. In addition, memantine has been shown to alter gene expression in adult rat brain [16] and modulate brain protein profiles in Down syndrome mouse model [17]. However, the proteomic alteration induced by memantine in AD-related mouse model has not been investigated yet. The aim of this study is to determine the protein profiles in different brain regions of the AD-related transgenic mouse model (3×Tg-AD) harboring the three mutations associated with familial AD (APP Swedish, MAPT P301L, and PSEN1 M146V). 3xTg-AD mice (8-month) were treated with memantine (5 mg/kg/bid) for 4 months with subsequent behavioral studies followed by detailed histopathological and molecular evaluation of the hippocampus and the cerebral cortex. The brain tissues were further analyzed by using quantitative proteomic analysis and bioinformatics to provide insights on the protein responses induced by memantine in the brain of AD mice.
MATERIALS AND METHODS
Animals and treatment
Female triple transgenic 3×Tg-AD mice harboring APPSwe, PSEN1M146V, and MAPTP301L transgenes (strain: B6; 129-Psen1tm1Mpm Tg [APPSwe, tauP301L]1Lfa/Mmjax) and female wild-type mice (WT) (strain: B6129SF2/J) were purchased from the Jackson Laboratory. Memantine was synthesized by Guangzhou Magpie Pharmaceuticals CO., LTD (Guangzhou, China). The mice were housed in groups of thirteen in big cages at room temperature with a 12:12 hour light/dark cycle. The 3×Tg-AD mice or WT mice (8-month old) were treated with memantine (5 mg/kg, bid, for 4 months) or with parallel volume of saline by intragastric administration, with 13 mice in each group. Cognitive capabilities of all animals were evaluated using behavioral tests. At the end of the behavioral tests, 6 mice from each group were sacrificed and the brain tissues were dissected and collected for proteomic study. All animals were conducted in accordance with the Guide for the Care and Use of Laboratory Animals as adopted and promulgated by the ethic committee of the Shenzhen Center for Disease Control and Prevention. All exertions were carried out to decrease the number of animals used and diminish animal suffering.
Behavioral tests
Step-down passive avoidance test
The step-down passive avoidance (SDA) test was performed according to the protocol described in our previous study [18]. Briefly, at training day, a single mouse was gently located on the grid floor, at the same time as electrical stimulation (36 V) was delivered through the grid floor for 5 min. On the day following completion of training, the mouse was gently put on the wooden platform and electrical stimulation was given for another 5 min. The memory was assessed by measuring the step-down latency recorded as the duration before the animal stepped down on the grid with 4 paws for the first time and the number of times the animal touching the grid floor with paws.
Novel object recognition test
The novel object recognition (NOR) test was performed according to the protocol described in our previous study [18]. Briefly, the single mouse was placed to the open-field arena containing two indistinguishable objects (A+A) and allowed to tour for 5 min once daily for 3 days. On day 4, a novel object was substituted for one of the old objects (A+B) in the open-field arena, and the mouse was placed to open-field arena to explore for 5 min as before. The behavior of the mouse was recorded by a video-tracking system. Discrimination index (DI) was used to evaluate the recognition ability, defined as the difference in exploration time between novel object (TN) and familiar object (TF), then dividing this value by the total amount of time spend exploring both object [DI=(TN−TF)/(TN+TF)*100.
Morris water maze test
Morris water maze (MWM) test was performed according to the protocol described in our previous study [18]. During training period, platform was located in the same position (one of four quadrants of the pool), and the mouse was placed into the pool facing the wall at one of the four start positions, and its movement was tracked by digital tracking system. The animal was immediately removed from the water when it located the platform. If the mouse did not located the platform after 60 s of swimming, it was gently guided to the platform or placed on the platform for an additional 15 s before being removed from the pool. The animal was tested in four trials per day with an inter-trial interval of approximately 30 min. The mice were trained for 5 days. The probe trial was performed on the sixth day following the last training session. During the probe trial, the platform was removed from the pool and the mouse was placed in the pool facing the wall from the diagonally opposite side of the platform. The mouse was allowed to swim freely for 2 min with digital movement-tracking recorded by computer software (SuperMaze+) before it was withdrawn from the pool.
Sample preparation
After behavioral testing, the mice were sacrificed by using intraperitoneal injections with 1% pentobarbital for collection of the brain tissues. Briefly, the hippocampus and the cerebral cortex were homogenized in ice-cold lysis buffer (8 M urea in PBS, pH 8.0, 1 mM protease inhibitor cocktail) using ultrasonic disruption system. Subsequently, the homogenates were centrifuged at 12,500×g for 20 min at 4℃, and the resulting supernatants were collected to a new tube. The protein concentration in the supernatant was determined by using BCA kit according to the manufacturer's instructions.
Tandem mass Tag (TMT) labeling
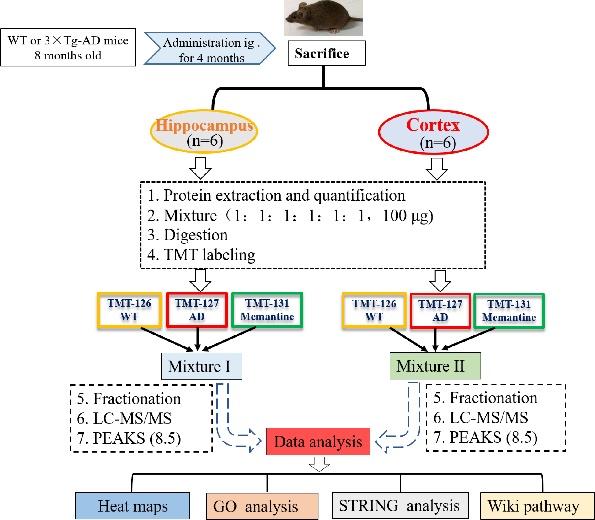
The flow diagram of proteomic research was shown in Fig. 1. For each group, 100 µg protein, either from the hippocampus or the cerebral cortex, was pooled from six animals. The pooled samples were treated with 300 µl of 10 mM DTT for 60 min at 55℃, followed by incubating with 300 µl of 25 mM IAA at room temperature in the dark for 60 min. Subsequently, the protein samples were diluted with PBS (final concentration of urea was 1.0 M) and digested via trypsin/Lys-C (1:25 w/w) overnight at 37℃ (Promega, WI, USA). On the second day, protein samples were heated to 65℃ and acidified with pure FA to achieve a final pH 1~2, and then centrifuged at 12,000×g for 15 min at 4℃ and collect supernatant. The samples were then desalted with reversed-phase column chromatography (Oasis HLB; Waters, MC, USA) and TMT isobaric labeled (Thermo Scientific, NJ, USA) according to manufacturer's instructions. Equal protein amounts (50 µg) derived from each group were labeled with different TMT labels: wild type (WT), TMT-126; 3×Tg-AD (AD) group, TMT-127; 3×Tg-AD+memantine (Memantine) group, TMT-131. The labeled peptides solution from WT, AD and Memantine group were mixed, dried and then fractionated into 8 fractions by high pH reversed-phase peptide fractionation kit (Thermo Scientific, NJ, USA). After that, each fraction peptide was dried using a vacuum centrifuge, then dissolved in 2% acetonitrile with 0.1% FA for LC-MS/MS analysis.
NanoLC-ESI-MS/MS analysis and database searches
The peptides fractions were examined on the AB SCIEX Triple-TOF 5600+ Mass Spectrometry (SCIEX, Concord, ON), equipped with an Eksigent nanoLC-Ultra™ 2D System. The fractions were loaded on Chrom XP C18 trap column (3 µm, 120 Å, 200 µm×0.5 mm, Eksigent) at a flow rate of 4 µl/min for 5 min and eluted through analytical column (75 µm×150 mm C18-3µm 120 Å, ChromXP, Eksigent) at a flow rate of 400 nl/min. The elution gradient was form 5% to 40 % solvent B (consisted of 2% ddH2O/98% acetonitrile/0.1% formic acid) in 90 min followed by 10 min at 95% solvent B and 20 min of re-equilibration with 5% solvent B. Raw data files generated by TripleTOF 5600+ Mass Spectrometry (SCIEX, Concord, ON) were converted to .mgf peaklists with PEAKS 8.5 software (Bioinformatics Solutions, Waterloo, Canada). For this procedure, we ran PEAKS 8.5 in identification mode against the UniProt-Mus musculus database (released in July 2017) with the following setting parameters: the parent mass error tolerance and fragment mass error tolerance were set at 30 ppm and 0.1 Da, respectively. The enzyme was set at trypsin. Fixed modification included the following: oxidation (M), deamidation (NQ), Pyro-glu from E, Pyro-glu from Q, Acetylation (Protein N-term) and maximum variable PTM per peptide: 3. A false discovery rate (FDR) less than 1.0% was selected for peptides only. Precise quantification of protein was expressed as protein ratio between samples, and set as group AD/WT, group memantine/WT and memantine/AD. The thresholds of up- and down-regulated ratio were set at 1.2 and 0.83, respectively.
The mass spectrometry proteomics data were archived into ProteomeXchange with identifier PXD012110 and PXD012110 (http://www.ebi.ac.uk/pride/archive).
Bioinformatics analysis
A first step for functional analysis of the altered protein list was to connect the protein accession with its associated gene ontology (GO) terms by using DAVID Bioinformatics resources version 6.8. The GO terms included biological process, cellular component and molecular function. Pathways enriched with altered proteins was determined by using KEGG pathway database (https://www.kegg.jp/kegg/). Heml 1.0 software and Graphpad 7.0 software were used to analyze the heat map. The Search Tool for the Retrieval of Interacting Genes (STRING; http://string-db.org) was used to evaluate protein-protein interaction (PPI) information. The interaction network was drawn by Cytoscape (3.6.0).
Statistical analysis
Data analysis of animal behavioral test was conducted by using GraphPad Prism 7.0 software with ANOVA and Tukey's HSD test.
RESULTS
Memantine ameliorated cognitive impairments of 3×Tg-AD mice
In the step-down passive avoidance (SDA) test, 3×Tg-AD (AD) mice exhibited shorter step-down latency when compared with wild type (WT) mice, and memantine significantly restored the step-down latency in AD mice (Fig. 2A). Similarly, Fig. 2B showed that AD mice made more errors than WT mice, and the number of errors was reduced when AD mice were treated with memantine. Results from the novel object recognition (NOR) test showed that AD mice had reduced abilities to recognize novel object (measured as Discrimination Index, DI), and memantine improved the DI although not reaching statistical significance (Fig. 2C).
In the Morris water maze (MWZ) test, AD mice took longer to reach the platform (escape latency) than WT mice during the training period, and memantine significantly decreased the escape latency of AD mice, with the effect most prominent on Day 5 (Fig. 3A). During the trial period, the platform was removed to evaluate if the mice after training would spend more time searching in the target quadrant than in the other quadrants. The trace records (Fig. 3B) showed that the swimming trajectory of AD mice in the target quadrant was less than those of WT mice and memantine treated AD mice. The AD mice took significantly much longer to find the target quadrant (Fig. 3C) and made less visits to the target quadrant (Fig. 3D). Both parameters could be significantly improved with the treatment of memantine in AD mice. Taken together, our data suggested that memantine was effective at improving the spatial memory of AD mice.
Memantine remodeled the hippocampal and cortical proteomes of 3×Tg-AD mice
Fig. 1 illustrated the experimental flowchart of proteomics analysis by using 6-plex TMT labeling quantitative proteomics strategy. Analysis of the hippocampal and cortical tissues of WT, 3×Tg-AD (AD), and memantine-treated AD mice showed that a total of 3301 proteins in the hippocampus and 3140 proteins in the cerebral cortex displayed altered expression with one or more unique peptides (false discovery rate (FDR) <1%). Among the altered proteins in the hippocampus, 233 were significantly modulated in memantine-treated AD mice (Fig. 4A), where 139 proteins were up-regulated and 94 proteins were down-regulated (with criteria ratio ≥1.2 or 0.83) (Table S-1). Further analysis identified 40 of the proteins with dramatic changes in levels (23 up-regulated, 17 down-regulated) (ratio ≥1.5 or <0.67) (Fig. 4B), and these proteins were involved in cytoskeleton organization, MAPK signaling pathway and axon guidance (Table S-2). PCTN (3.58), ATAXIN-2 (2.37), TNIK (1.98), NOL3 (1.78) were the most up-regulated proteins, and DPP9 (0.6), BRAF (0.59), FLNA (0.57), MARK2 (0.44) were the most down-regulated proteins (Table S-2). In the cerebral cortex, 342 proteins were significantly modulated in memantine-treated AD mice (80 up-regulated, 262 down-regulated) (ratio ≥1.2 or <0.83) (Fig. 4C, Table S-3). Among of them, 50 proteins had dramatic changes in expression levels (fold change ≥ 1.5 or <0.67) as shown in Fig. 4D (Table S-4), and they were related to cytoskeleton organization, mitochondrial function, oxidative reduction and neuron development. The most up-regulated proteins were MPPB (2.69), PCNT (2.62), Myelin (1.62) and the most down-regulated proteins were Dynamin-2 (0.63), BRAF (0.53), MOES (0.41). These proteins are all highly associated with AD.
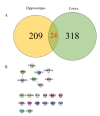
We found that only 24 proteins, including MPPB, MOES, PCNT and BRAF, were significantly changed in both hippocampus and cerebral cortex, as shown in Fig. 5A and Table 1. Among the 24 proteins, only 8 proteins had interaction with each other as shown in Fig. 5B. Some of the altered proteins under the influence of memantine showed opposite expression patterns (Table 1). MBBP, RAB8A and UFM1 were down-regulated (ratio memantine/AD <0.83, blue number in Table 1) in the hippocampus but upregulated (ratio memantine/AD>1.2, red number in Table 1) in the cerebral cortex. On the other hand, MFR1L, PDE1C, U520, NB5R1, PCBP3, ADT4, CRTC1 and MOES were up-regulated (ratio memantine/AD>1.2, green number in Table 1) in the hippocampus while they were down-regulated in the cerebral cortex (ratio memantine/AD <0.83, purple in Table 1).
Bioinformatics analysis of altered proteins
To investigate the functional significance of the altered proteins, we performed a functional classification by using DAVID bioinformatics tool (version 6.8) according to Gene Ontology (GO) terms (biological process, molecular function and cellular component) as shown in the Fig. 6. In the hippocampus, the most enriched terms of biological process were cellular macromolecular complex assembly, intracellular signaling cascade and small GTPase mediated signal transduction. The most enriched terms of cellular component for the hippocampal proteins were cytoskeleton, non-membrane-bounded organelle and synapse. The most over-represented terms of molecular function for the hippocampal proteins were nucleotide binding, GTP binding and guanyl nucleotide binding. For the cerebral cortical proteins, the most over-represented terms of biological process were establishment of protein localization, protein localization and protein transport. The most over-represented terms of cellular component were mitochondrion, synapse and cytoskeleton. The most over-represented terms of molecular function were nucleotide binding, purine nucleotide binding and ribonucleotide binding. KEGG pathway analysis was used to present these characteristics. For the hippocampal proteins, the highly enriched terms included longterm potentiation, insulin signaling pathway, amino sugar and nucleotide sugar metabolism, vascular smooth muscle contraction and regulation of actin cytoskeleton. For the cerebral cortical proteins, the highly enriched terms included long-term potentiation, glycerolipid metabolism, calcium signaling pathway, insulin signaling pathway and MAPK signaling pathway.
To further evaluate the relationships among the differentially expressed proteins (ratio ≥1.2 or <0.83) induced by memantine treatment in 3×Tg-AD mice, protein-protein interaction (PPI) networks were performed using STRING database and interpreted with Cytoscape 3.6.0. In the hippocampus and the cerebral cortex, more than half of the differentially expressed proteins were connected with each other, many of which were involved in AD pathology. In the hippocampus (Fig. 7A), the most represented pathways were ErbB signaling pathway, Alzheimer's disease and regulation of cytoskeleton. In the cerebral cortex (Fig. 7B), the pattern of modulation was more complex than in the hippocampus. This included electron transfer activity, Alzheimer's disease, ribosome, axon guidance, calcium signaling pathway, MAPK signaling pathway, regulation of cytoskeleton and vesicle-mediated transport. The data implicated that memantine might modulate multiple pathways to protect against neuronal death.
To further understand the signaling pathways affected by memantine treatment, we conducted Wiki pathway analysis using Cytoscape 3.6.0 software. We imported the information of all the proteins identified in the hippocampus or the cerebral cortex into Cytoscape 3.6.0 software for mapping the cytoplasmic ribosomal proteins pathway in an integrated analysis (Fig. 8). We found that most of the ribosomal protein subunits in the hippocampus or the cerebral cortex were affected by memantine treatment. In the hippocampus, only small (40 S) subunit protein Rps 12 was significantly down-regulated. However, in the cerebral cortex, 60 S subunit protein Rpl32 and Rpl35 were significantly down-regulated, and the 40 S subunit protein Rps6ka2 was significantly upregulated. The data indicated that memantine might have a greater effect on the cerebral cortex than the hippocampus.
DISCUSSION
Here, we evaluated the therapeutic effects of memantine against cognitive declines in 3×Tg-AD mice and characterized the proteome profiles in the hippocampus and the cerebral cortex. Our results from behavioral tests indicated that memantine was effective for improving cognitive abilities in 3×Tg-AD mice, including learning and memory (SDA test), recognition memory (NOR test), and spatial navigation and reference memory (MWM test). Our results from proteomics profiling showed that memantine significantly regulated proteins relevant to Alzheimer's disease (AD), including proteins for axon guidance, cytoskeleton, and ribosomal proteins, and modulated proteins connected to calcium and MAPK signaling pathways. To the best of our knowledge, this is the first study to report a comprehensive proteomic analysis of the hippocampus and the cerebral cortex under the influence of memantine treatment in AD-related mouse model. In particular, we observed a significant down-regulation of microtubule-affinity regulating kinase 2 (MARK2) in memantine-treated AD mice, with ratio of 0.437 and 0.759 in the hippocampus and the cerebral cortex, respectively.
MARK2 is one of the four MARK isoforms originally discovered by its ability to phosphorylate the Lys-X-Gly-Ser (KXGS) motifs in the repeat domains of microtubule-associated proteins tau (MAPT), MAP2, and MAP4, and thereby to regulate microtubule dynamics in neurons [19,20]. MARKs also display diverse functions in neuronal polarity, transport, cell-cycle control and cytoskeleton, which play a prominent role in neurodegenerative ‘tauopathies’ such as AD [21,22]. The formation of neurofibrillary tangles (NFTs) is one of the major hallmarks in the post-mortem brain of AD [23]. Tau binds to and stabilizes microtubules (MTs) in the neurons, especially in neuronal axons [23]. When tau undergoes abnormal hyper-phosphorylation in the AD brain, its affinity for MTs is reduced and it accumulates as NFTs, contributing to the development of synaptic dysfunction and neuronal loss [23,24]. Studies have reported that the elevated interactions between MARK2 and tau were observed in post-mortem AD brains [25]. It has been demonstrated that active MARK2 interacted with tau and phosphorylated tau at Ser262 in stably transfected cell line, while the interaction between MARK2 and tau was significantly reduced when MARK2 was inhibited by protein kinase inhibitor [25]. The overexpression of MARK2 led to hyperphosphorylation of MAPs and disturbance in microtubule array, resulting in cellular morphological changes and cell death in Chinese hamster ovary (CHO) cells [19,26], while knockdown of MARK2 expression using siRNA contributed to the formation of multiple axon like neurites and elongation of axons in hippocampal neurons [26,27]. One study reported that MARK2 and PTEN-induced kinase 1 (PINK1) were co-localized with mitochondrial and regulate their transport, and MARK2 was identified as an upstream regulator of PINK1 [28].
The activation of NMDA receptors (NMDAR) leads to calciuminflux and the activation of downstream effectors such as CaM-KII, PI3K, ERK and PKA. Previous studies have reported that NMDAR activated protein kinase A (PKA) which phosphorylated MARK2 [29]. MARK2 could regulate microtubule stability, neurite outgrowth, and early phosphorylation of tau in AD [26]. The augmented interactions between MARK2 and tau in AD brain suggest that MARK2 plays an important role in the early phosphorylation of tau in AD, indicating it as a potential therapeutic target. More recently, a series of selective MARK inhibitors have been synthesized and examined for potential treatment for AD [23]. Our data provided further evidence of the roles of MARK2 in AD pathogenesis, and demonstrated the association between MAPK2 and memantine therapeutic effects in AD for the first time.
SUPPLEMENTARY MATERIALS
Table S-1: 233 proteins in the hippocampus that were significantly changed by memantine treatment.
Table S-2: 40 proteins with dramatic changes in levels in the hippocampus by memantine treatment.
Table S-3. 342 proteins in the cerebral cortex that were significantly changed by memantine treatment.
Table S-4. 50 proteins with dramatic changes in levels in the cerebral cortex by memantine treatment.
en-28-390-s001.pdfFigures
{kind=link}

{kind=link}

{kind=link}

{kind=link}
{kind=link}

{kind=link}

{kind=link}

{kind=link}
Tables
{kind=link}
GN means Gene name.
References
- Reisberg B, Doody R, Stöffler A, Schmitt F, Ferris S, Möbius HJ, Memantine Study Group. Memantine in moderate-to-severe Alzheimer's disease. N Engl J Med 2003;348:1333-1341.

- Du X, Wang X, Geng M. Alzheimer's disease hypothesis and related therapies. Transl Neurodegener 2018;7:2.
- Martinez-Coria H, Green KN, Billings LM, Kitazawa M, Albrecht M, Rammes G, Parsons CG, Gupta S, Banerjee P, LaFerla FM. Memantine improves cognition and reduces Alzheimer's-like neuropathology in transgenic mice. Am J Pathol 2010;176:870-880.
- Scholtzova H, Wadghiri YZ, Douadi M, Sigurdsson EM, Li YS, Quartermain D, Banerjee P, Wisniewski T. Memantine leads to behavioral improvement and amyloid reduction in Alzheimer's-disease-model transgenic mice shown as by micromagnetic resonance imaging. J Neurosci Res 2008;86:2784-2791.
- Yiannopoulou KG, Papageorgiou SG. Current and future treatments for Alzheimer's disease. Ther Adv Neurol Disord 2013;6:19-33.
- Di Iorio G, Baroni G, Lorusso M, Montemitro C, Spano MC, di Giannantonio M. Efficacy of memantine in schizophrenic patients: a systematic review. J Amino Acids 2017;2017:7021071.
- Johnson JW, Kotermanski SE. Mechanism of action of memantine. Curr Opin Pharmacol 2006;6:61-67.
- Danysz W, Parsons CG. Alzheimer's disease, β-amyloid, glutamate, NMDA receptors and memantine--searching for the connections. Br J Pharmacol 2012;167:324-352.
- Sekiguchi K, Sato M, Yokoyama MK, Sato T, Tsutiya A, Omoteyama K, Arito M, Suematsu N, Kato T, Kurokawa MS. Effects of memantine on the growth and protein profiles of neuroblastoma cells. Integr Mol Med 2018;5:1-8.
- Rudy CC, Hunsberger HC, Weitzner DS, Reed MN. The role of the tripartite glutamatergic synapse in the pathophysiology of Alzheimer's disease. Aging Dis 2015;6:131-148.
- Wang X, Blanchard J, Grundke-Iqbal I, Iqbal K. Memantine attenuates Alzheimer's disease-like pathology and cognitive impairment. PLoS One 2015;10:e0145441.
- Song MS, Rauw G, Baker GB, Kar S. Memantine protects rat cortical cultured neurons against beta-amyloid-induced toxicity by attenuating tau phosphorylation. Eur J Neurosci 2008;28:1989-2002.
- De Felice FG, Velasco PT, Lambert MP, Viola K, Fernandez SJ, Ferreira ST, Klein WL. Aβ oligomers induce neuronal oxidative stress through an
N -methyl-D-aspartate receptor-dependent mechanism that is blocked by the Alzheimer drug memantine. J Biol Chem 2007;282:11590-11601.
- Nagakura A, Shitaka Y, Yarimizu J, Matsuoka N. Characterization of cognitive deficits in a transgenic mouse model of Alzheimer's disease and effects of donepezil and memantine. Eur J Pharmacol 2013;703:53-61.
- Alley GM, Bailey JA, Chen D, Ray B, Puli LK, Tanila H, Banerjee PK, Lahiri DK, Lahiri DK. Memantine lowers amyloid-beta peptide levels in neuronal cultures and in APP/PS1 transgenic mice. J Neurosci Res 2010;88:143-154.
- Marvanová M, Lakso M, Wong G. Identification of genes regulated by memantine and MK-801 in adult rat brain by cDNA microarray analysis. Neuropsychopharmacology 2004;29:1070-1079.
- Ahmed MM, Dhanasekaran AR, Block A, Tong S, Costa AC, Gardiner KJ. Protein profiles associated with context fear conditioning and their modulation by memantine. Mol Cell Proteomics 2014;13:919-937.
- Zhou X, Xiao W, Su Z, Cheng J, Zheng C, Zhang Z, Wang Y, Wang L, Xu B, Li S, Yang X, Pui Man Hoi M. Hippocampal proteomic alteration in triple transgenic mouse model of Alzheimer's disease and implication of PINK 1 regulation in donepezil treatment. J Proteome Res 2019;18:1542-1552.
- Drewes G, Ebneth A, Preuss U, Mandelkow EM, Mandelkow E. MARK, a novel family of protein kinases that phosphorylate microtubule-associated proteins and trigger microtubule disruption. Cell 1997;89:297-308.
- Drewes G. MARKing tau for tangles and toxicity. Trends Biochem Sci 2004;29:548-555.
- Matenia D, Mandelkow EM. Emerging modes of PINK1 signaling: another task for MARK2. Front Mol Neurosci 2014;7:37.
- Matenia D, Mandelkow EM. The tau of MARK: a polarized view of the cytoskeleton. Trends Biochem Sci 2009;34:332-342.
- Katz JD, Haidle A, Childers KK, Zabierek AA, Jewell JP, Hou Y, Altman MD, Szewczak A, Chen D, Harsch A, Hayashi M, Warren L, Hutton M, Nuthall H, Su HP, Munshi S, Stanton MG, Davies IW, Munoz B, Northrup A. Structure guided design of a series of selective pyrrolopyrimidinone MARK inhibitors. Bioorg Med Chem Lett 2017;27:114-120.
- Lee VM, Brunden KR, Hutton M, Trojanowski JQ. Developing therapeutic approaches to tau, selected kinases, and related neuronal protein targets. Cold Spring Harb Perspect Med 2011;1:a006437.
- Gu GJ, Wu D, Lund H, Sunnemark D, Kvist AJ, Milner R, Eckersley S, Nilsson LN, Agerman K, Landegren U, Kamali-Moghaddam M. Elevated MARK2-dependent phosphorylation of Tau in Alzheimer's disease. J Alzheimers Dis 2013;33:699-713.
- Deng SS, Wu LY, Wang YC, Cao PR, Xu L, Li QR, Liu M, Zhang L, Jiang YJ, Yang XY, Sun SN, Tan MJ, Qian M, Zang Y, Feng L, Li J. Protein kinase A rescues microtubule affinity-regulating kinase 2-induced microtubule instability and neurite disruption by phosphorylating serine 409. J Biol Chem 2015;290:3149-3160.
- Chen YM, Wang QJ, Hu HS, Yu PC, Zhu J, Drewes G, Piwnica-Worms H, Luo ZG. Microtubule affinity-regulating kinase 2 functions downstream of the PAR-3/PAR-6/atypical PKC complex in regulating hippocampal neuronal polarity. Proc Natl Acad Sci U S A 2006;103:8534-8539.
- Matenia D, Hempp C, Timm T, Eikhof A, Mandelkow EM. Microtubule affinity-regulating kinase 2 (MARK2) turns on phosphatase and tensin homolog (PTEN)-induced kinase 1 (PINK1) at Thr-313, a mutation site in Parkinson disease: effects on mitochondrial transport. J Biol Chem 2012;287:8174-8186.
- Bernard LP, Zhang H. MARK/Par 1 kinase is activated downstream of NMDA receptors through a PKA-dependent mechanism. PLoS One 2015;10:e0124816.