Articles

Article Tools
 View Full Text View Full Text |
 Abstract Abstract |
 Article as PDF Article as PDF |
 Print this Article Print this Article |
 Pubmed Pubmed |
 PMC PMC |
 PubReader PubReader |
 Export to Citation Export to Citation |
 Email Alerts Email Alerts |
 Open Access Open Access |
Supplementary
Share this article on :



Stats or Metrics
Article
Original Article
Exp Neurobiol 2019; 28(4): 516-528
Published online August 31, 2019
https://doi.org/10.5607/en.2019.28.4.516
© The Korean Society for Brain and Neural Sciences
Spinal Nitric Oxide Synthase Type II Increases Neurosteroid-metabolizing Cytochrome P450c17 Expression in a Rodent Model of Neuropathic Pain
Sheu-Ran Choi1, Alvin J Beitz2 and Jang-Hern Lee1*
1Department of Veterinary Physiology, BK21 PLUS Program for Creative Veterinary Science Research, Research Institute for Veterinary Science and College of Veterinary Medicine, Seoul National University, Seoul 08826, Korea, 2Department of Veterinary and Biomedical Sciences, College of Veterinary Medicine, University of Minnesota, St. Paul, MN 55108, USA
Correspondence to: *To whom correspondence should be addressed.
TEL: 82-2-880-1272, FAX: 82-2-885-2732
e-mail: jhl1101@snu.ac.kr
This is an Open Access article distributed under the terms of the Creative Commons Attribution Non-Commercial License(http://creativecommons.org/licenses/by-nc/4.0) which permits unrestricted non-commercial use, distribution, andreproduction in any medium, provided the original work is properly cited.
Abstract
We have previously demonstrated that the neurosteroid dehydroepiandrosterone sulfate (DHEAS) induces functional potentiation of
Graphical Abstract

Keywords: Nitric oxide synthase type II, Cytochrome P450c17, Dehydroepiandrosterone sulfate, Phosphorylation, Neuropathic pain
INTRODUCTION
Several lines of evidence indicate that some steroids, called neurosteroids, are synthesized in the nervous system in an independent manner from the steroidogenic organs and profoundly influence neuronal excitability [1, 2]. One of these neurosteroids, dehydroepiandrosterone sulfate (DHEAS), has been found within the central nervous system of several mammalian species [3, 4]. DHEAS is a negative modulator of the gamma-aminobutyric acid type A (GABAA) receptors and a positive activator of both sigma-1 receptors and
Based on data from experimental studies, there is increasing support for the involvement of both peripheral and central mechanisms in chronic neuropathic pain. Nitric oxide (NO) is a free gaseous signaling molecule synthesized by different isoforms of nitric oxide synthase (NOS) and has been linked with the development and maintenance of neuropathic pain [10–13]. Among the isoforms of NOS, nitric oxide synthase type II (NOS-II), which is also known as inducible NOS, has been demonstrated to produce a large amount of NO, thus, its increased expression plays an important role in the facilitation of inflammatory reactions and neurotoxicity [12, 14]. It has been suggested that local activation of the NOS-II/NO system plays an important role in the pathogenesis of peripheral neuropathy and the development of chronic pain [15]. In the peripheral nervous system, NOS-II was detected locally in the paw 6 h after Complete Freund’s Adjuvant (CFA) injection. Inhibition of iNOS significantly reversed CFA-induced hypersensitivity to pain and edema in a dose-dependent manner demonstrating a role for peripherally-expressed NOS-II in pain conditions with an inflammatory component [16]. Although NOS-II regulates the gene expression of a variety of proteins in the central nervous system, resulting in modulation of the synaptic environment and neuronal excitability in the spinal cord and brain [14], the potential role of NOS-II activation on neurosteroid signaling induced by P450c17 is poorly understood, particularly in the case of spinal nociceptive signaling transmission.
The main purpose of this study was to investigate whether spinal NOS-II modulates the expression of P450c17 and GluN1 phosphorylation, ultimately leading to the development of chronic pain in a rat model of neuropathic pain. In this regard, we examined whether: (1) sciatic nerve injury increases NOS-II expression and NO levels in the lumbar spinal cord dorsal horn of CCI rats; (2) inhibition of spinal NOS-II with L-NIL suppresses the development of neuropathic pain, GluN1 phosphorylation, and spinal P450c17 expression; and (3) co-administration of DHEAS together with L-NIL restores the CCI-induced development of neuropathic pain and GluN1 phosphorylation that were significantly reduced by L-NIL administration alone.
MATERIALS AND METHODS
Experimental animals and surgery
Male Sprague-Dawley rats (180~200 g) were purchased from the Laboratory Animal Center of Seoul National University (Seoul, Republic of Korea). Animals had free access to food and water and were kept in temperature and light controlled rooms (23±2°C, 12/12h light/dark cycle) for 3 days prior to the beginning of the experiment. The experimental protocols for animal usage were reviewed and approved by the SNU Institutional Animal Care and Use Committee and were consistent with the Guide for the Care and Use of Laboratory Animals published by the US National Institutes of Health (NIH publication No. 96-01, revised 1996).
A chronic constriction injury (CCI) of the common sciatic nerve was performed according to the method described by Bennett and Xie [17]. Briefly, rats were anesthetized with 3% isoflurane in a mixture of N2O/O2 gas. The right sciatic nerve was exposed and 4 loose ligatures of 4-0 chromic gut were placed around the nerve and spaced apart from each other by 1.0- to 1.5-mm separations. Sham surgery was performed by exposing the sciatic nerve in the same manner, but without ligating the nerve. After surgery, animals recovered in clear plastic cages at 27°C with a thick layer of sawdust bedding.
Drugs and administration
The following drugs were used: L-N6-(1-Iminoethyl)lysine dihy-drochloride (L-NIL, an NOS-II inhibitor; 6, 20, 60 nmol); dehydroepiandrosterone sulfate (DHEAS; 3, 10 nmol). L-NIL and DHEAS were purchased from Sigma–Aldrich (St. Louis, MO, USA). The doses of L-NIL were selected based on the results of preliminary experiments and the doses of DHEAS were selected based on the doses used in a previous study from our laboratories [8]. L-NIL was dissolved in physiological saline and DHEAS was dissolved in 1% DMSO in physiological saline. The injection volume was 20 μl. Drugs were administered intrathecally twice a day on postoperative days 0~5, during the induction phase of neuropathic pain development.
Intrathecal drug administration was performed using a 50 μl Hamilton syringe connected to a 27-gauge needle as previously described [18]. Rats were briefly anesthetized with 3% isoflurane in a mixture of N2O/O2 gas to prevent any handling-induced stress and to allow more accurate injection of drugs. The rat was held tightly between the thumb and middle finger at the level of the both iliac crests, and the fifth lumbar spinous process was palpated with the index finger. The needle was inserted through the vertebral column into the L5–6 intervertebral space and successful insertion of the needle into the intrathecal space was determined by a tail flick response. Each drug was slowly injected over a 10 second period. Then, the needle was carefully removed from the spinal cord. The drug control groups received an identical injection of vehicle.
Behavioral assessment
Pain behavioral tests were performed on the ipsilateral (injured) hind paw 1 day before surgery on all animals to obtain normal baseline values of paw withdrawal responses to mechanical and thermal stimulation. Then, animals were randomly assigned to experimental and control groups. Animals were tested again at 1, 3, 4, 5, 7, 9, 14, 21 and 28 days following CCI surgery. All behavioral analyses were performed blindly.
To assess nociceptive responses to innocuous mechanical stimuli (mechanical allodynia), we measured paw withdrawal response frequency (PWF) by using a von Frey filament with a force of 2.0 g (North Coast Medical, Morgan Hill, CA) as described in a previous study from our laboratories [18]. Rats were placed in acrylic chambers on a wire mesh floor and allowed to habituate before testing. A von Frey filament was applied to the plantar surface of each hind paw for a 3 sec period before removal and we subsequently recorded whether there was a withdrawal of the hind limb to the filament. The filament was applied 10 times to the hind paw with a 10 sec interval between each application. Then, the number of paw withdrawal responses was counted and the results of mechanical behavioral testing in the hind paw were expressed as a percent withdrawal response frequency, which represented the percentage of paw withdrawals out of the maximum of 10.
To assess nociceptive responses to noxious heat stimuli (thermal hyperalgesia), we measured the paw withdrawal response latency (PWL, s) using a plantar analgesia meter (Model 390, IITC Life Science Inc., Woodland Hills, CA, USA) as previously described by Hargreaves et al. with minor modifications [19]. Rats were placed in acrylic cylinders on a glass floor and allowed to habituate before testing. A radiant heat source was positioned under the floor beneath the hind paw and then the withdrawal latency was measured. The test was duplicated in the ipsilateral hind paw, and the mean withdrawal latency was calculated. A cutoff latency in the absence of a response was set at 20 s to prevent tissue damage.
Measurement of nitric oxide in the spinal cord
Nitric oxide (NO) was determined in the spinal cord dorsal horns from the lumbar enlargement using a nitric oxide detection kit (cat# ADI-917-020, Enzo Life sciences Inc., USA) as described previously [20]. This nitric oxide (total) detection kit is based on the enzymatic conversion of nitrate to nitrite by the enzyme nitrate reductase, followed by the Griess reaction to form a colored azo dye product. Advantages of this method include a strong literature background, numerous commercially available reagent kits and wide availability of infrastructure. Animals were deeply anesthetized with 3% isoflurane in a mixture of N2O/O2 gas on day 5 post-surgery and perfused transcardially with calcium-free Tyrode’s solution. The isolated spinal cords were homogenized in PBS by sonication. Homogenates were subsequently centrifuged at 400 g for 10 min at 4°C and, then, the supernatant was used for nitric oxide detection following the manufacturer’s recommendation.
Western blot assay
The Western blot assay was performed as described previously [18]. Animals were deeply anesthetized with 3% isoflurane in a mixture of N2O/O2 gas on day 5 post-surgery and perfused transcardially with calcium-free Tyrode’s solution. The ipsilateral spinal cord dorsal horns from the lumbar enlargement were homogenized in lysis buffer (20 mM Tris-HCl, 10 mM EGTA, 2 mM EDTA, pH 7.4 and proteinase inhibitors) containing 1% Triton X-100. Homogenates were subsequently centrifuged at 15,000 rpm for 40 min at 4°C and, then, the supernatant was used for Western blot analysis.
The protein concentration was estimated by the Bradford dye assay (Bio-Rad Laboratories). Spinal cord homogenates (25 μg protein) were separated using 10% SDS-polyacrylamide gel electrophoresis and transferred to nitrocellulose membrane. After the blots had been washed with TBST (10 mM Tris-HCl, pH 7.6, 150 mM NaCl and 0.05% Tween-20), the membranes were blocked with 5% skimmed milk for 1 h at RT and incubated at 4°C overnight with a primary antibody specific for NOS-II (rabbit polyclonal anti-NOS2 antibody, 1:1,000, cat# sc-651, Santa Cruz Biotechnology Inc.), PKC-dependent pGluN1 (rabbit polyclonal anti-pGluN1 Ser896 antibody, 1:1,000, cat# ABN88, Millipore Co., USA; The antibody-specificity information available from Millipore Corporation states that this antibody recognizes GluN1 when phosphorylated at serine 896 and that the immunogen was a KLH-conjugated linear peptide corresponding to rat GluN1 phosphorylated at serine 896), PKA-dependent pGluN1 (rabbit polyclonal anti-pGluN1 Ser897 antibody, 1:1,000, cat# ABN99, Millipore Co., USA; The antibody-specificity information available from Millipore Corporation states that this antibody recognizes GluN1 when phosphorylated at serine 897 and the immunogen was a KLH-conjugated linear peptide corresponding to rat GluN1 phosphorylated at serine 897), P450c17 (rabbit monoclonal anti-cytochrome P450 17A1 antibody, 1:1,000, cat# ab125022, Abcam plc.) or β-actin (mouse monoclonal anti-β-actin antibody, 1:5,000, cat# sc-47778, Santa Cruz Biotechnology Inc.). After washing with TBST, membranes were incubated for 4 h at 4°C with horseradish peroxidase-conjugated anti-rabbit or anti-mouse antibody (1:10,000, Santa Cruz Biotechnology Inc.). The bands were visualized with enhanced chemiluminescence (Amersham Biosciences). The positive pixel area of specific bands was measured using ImageJ software (ImageJ 1.45s; National Institutes of Health, USA) and normalized against the corresponding β-actin loading control bands. The mean value of control groups was set at 100%. Thus, the % change relative to the mean value of control groups was then calculated in each group.
Quantitative real-time PCR
Quantitative real-time PCR was performed as described previously [9]. Animals were deeply anesthetized with 3% isoflurane in a mixture of N2O/O2 gas on day 5 post-surgery and perfused transcardially with calcium-free Tyrode’s solution. The ipsilateral spinal cord dorsal horns from the lumbar enlargement were homogenized in Buffer RL, and RNA samples were extracted using a commercial RNA extraction kit (cat# 9767, TaKaRa, Otsu, Shiga, Japan). Then, 1 μg of RNA was reverse-transcribed with a reverse transcription-PCR premix (cat# 25081, iNtRON Biotechnology, Seongnam, Korea). Reverse transcription was performed for 1 h at 45°C followed by 5 min at 95°C. The cDNA samples were amplified with the specific mRNA primers and a TBTM Green Premix Ex TaqTM (cat# RR420A, TaKaRa). Quantitative real-time PCR was performed as follows: 10 min at 95°C for DNA polymerase activation and 50 cycles of 15 s at 94°C, 15 s at 58°C, and 30 s at 72°C. The identity and specificity of the amplified PCR product was validated by melting curve analysis. The relative expression of P450c17 mRNA was quantified with double delta Ct analysis, and the data were normalized with the expression of GAPDH mRNA. The quantitative real-time PCR experiment was duplicated for each sample and the mean mRNA level was calculated. The sequences of the mRNA primers used in this study are described in Table 1.
Immunohistochemistry
Rats were deeply anesthetized with 3% isoflurane in a mixture of N2O/O2 gas at day 5 post-CCI surgery and perfused transcardially with calcium-free Tyrode’s solution and subsequently with fixative containing 4% paraformaldehyde in 0.1 M phosphate buffer (pH 7.4). The spinal cords were collected after perfusion, postfixed in the identical fixative for 2 h at RT and then placed in 30% sucrose in PBS (pH 7.4) at 4°C. Serial transverse sections (40 μm) of the L4–5 spinal cord were cut using a cryostat (Leica CM1520, Leica Biosystems, Germany). Spinal tissue sections were washed with PBS and incubated for 2 days at 4°C with a primary antibody specific for NOS-II (rabbit polyclonal anti-NOS2 antibody, 1:1,000, cat# sc-651, Santa Cruz Biotechnology Inc.) or Iba-1 (goat polyclonal anti-Iba-1 antibody, 1:500, cat# ab5076, Abcam plc.). The primary antibodies were detected by incubating the tissue in Alexa Fluor® 568 donkey anti-rabbit antibody (1:400, Invitrogen) or Alexa Fluor® 488 donkey anti-goat antibody (1:400, Life Technologies) for 90 min at RT. Tissue sections were mounted on slides and visualized with a confocal microscope (Nikon Eclipse TE2000-E, Nikon, Japan).
Statistical analysis
Data are expressed as the mean±SEM. Statistical analyses were performed using Prism 5.0 (Graph Pad Software, San Diego, USA). Repeated measures two-way ANOVA was performed to determine differences in the behavioral data and this was followed by a Bonferroni multiple comparison test for
RESULTS
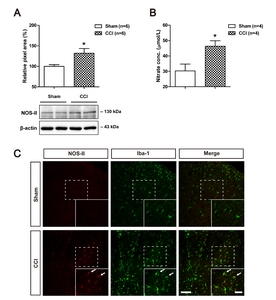
Sciatic nerve injury increases the expression of NOS-II in microglial cells and NO levels in the lumbar spinal cord dorsal horn of CCI rats
To determine whether chronic constriction injury (CCI) of the sciatic nerve induces a significant change in the expression of NOS-II and NO levels in the lumbar spinal cord dorsal horn, we examined the expression of NOS-II using Western blot analysis and measured nitrate concentration using an NO detection kit (a diazotization assay). Sciatic nerve injury significantly increased the expression of NOS-II (Fig. 1A) and NO levels (Fig. 1B) in the lumbar spinal cord dorsal horn at day 5 post-surgery as compared to that of the sham group (Fig. 1A and 1B; *p<0.05 vs. Sham). In addition, the results of immunohistochemistry analysis show that sciatic nerve injury increased NOS-II immunostaining, which is co-localized in Iba-1-positive microglial cells (Fig. 1C).
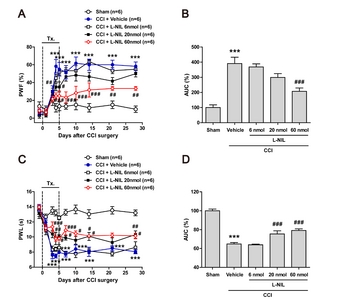
I.t. administration of L-NIL suppresses the CCI-induced development of neuropathic pain in rats
To verify whether the CCI-induced development of neuropathic pain is induced by activation of NOS-II, we intrathecally injected the NOS-II inhibitor, L-NIL during the induction phase of neuropathic pain. Sciatic nerve injury increased the paw withdrawal frequency (PWF, %) to innocuous mechanical stimuli (mechanical allodynia) (Fig. 2A; ***p<0.001 vs. Sham). The increase in PWF was significantly different at 4 days and this difference was sustained for 28 days post-surgery as compared to the sham group. Repeated daily intrathecal (i.t.) administration of L-NIL (6, 20 and 60 nmol) during the induction phase of neuropathic pain (from days 0~5 post-surgery) dose-dependently attenuated the CCI-induced development of mechanical allodynia as compared with vehicle-treated CCI rats (Fig. 2A; #p<0.05, ##p<0.01, ###p<0.001 vs. vehicle-treated group). In addition, area under curve (AUC, %) data analysis showed a significant analgesic effect of L-NIL on the development of mechanical allodynia in neuropathic rats (Fig. 2B; ***p<0.001 vs. Sham, ###p<0.001 vs. vehicle-treated group).
Sciatic nerve injury also decreased the paw withdrawal latency (PWL, s) to noxious heat stimulation (thermal hyperalgesia) as compared to the sham group, and this decrease was dose-dependently inhibited by i.t. administration of L-NIL (6, 20 and 60 nmol) during the induction phase of neuropathic pain (from days 0~5 post-surgery) as compared with vehicle-treated CCI rats (Fig. 2C; ***p<0.001 vs. Sham, #p<0.05, ##p<0.01, ###p<0.001 vs. vehicle-treated group). In addition, the AUC (%) data analysis showed a significant analgesic effect of L-NIL on the development of thermal hyperalgesia in neuropathic rats (Fig. 2D; ***p<0.001 vs. Sham, ###p<0.001 vs. vehicle-treated group).
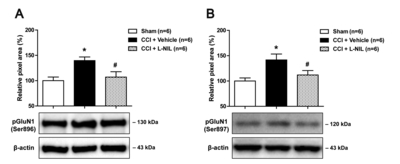
I.t. administration of L-NIL suppresses the CCI-induced increase in PKC- and PKA-dependent GluN1 phosphoryla-tion in the lumbar spinal cord dorsal horn of rats
In order to determine whether NOS-II modulates the functional activation of NMDA receptors, we examined the changes in the PKC- and PKA-dependent phosphorylation of the NMDA receptor GluN1 subunit (pGluN1) using Western blot analysis. Sciatic nerve injury increased PKC-dependent pGluN1 at the Ser896 site (Fig. 3A) and PKA-dependent pGluN1 at the Ser897 site (Fig. 3B) in the lumbar spinal cord dorsal horn at day 5 post-surgery as compared to that of the sham group (Fig. 3; *p<0.05 vs. Sham). Intrathecal administration of L-NIL (60 nmol) during the induction phase of neuropathic pain (from days 0~5 post-surgery) significantly decreased not only the PKC-dependent pGluN1 at the Ser896 site (Fig. 3A) but also the PKA-dependent pGluN1 at the Ser897 site (Fig. 3B) in CCI rats (Fig. 3; #p<0.05 vs. vehicle-treated group). These results demonstrate that activation of NOS-II increases the PKC- and PKA-dependent phosphorylation of GluN1 in the lumbar spinal cord dorsal horn, which may lead to the potentiation of NMDA receptor function and the development of neuropathic pain following CCI.
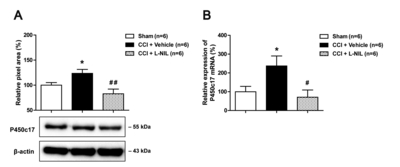
I.t. administration of L-NIL suppresses the increases in protein and mRNA levels of P450c17 in the lumbar spinal cord dorsal horn of CCI rats
Next, we examined the effect of the NOS-II inhibitor, L-NIL on the protein and mRNA levels of the key DHEA-synthesizing enzyme, cytochrome P450c17 in the lumbar spinal cord dorsal horn using Western blot analysis and quantitative real-time PCR, respectively. Sciatic nerve injury significantly increased the protein level of P450c17 in the lumbar spinal cord dorsal horn at day 5 post-surgery as compared to that of the sham group (Fig. 4A; *p<0.05 vs. Sham), and this increase was significantly inhibited by i.t. administration of L-NIL (60 nmol) during the induction phase of neuropathic pain (from days 0~5 post-surgery) in CCI rats (Fig. 4A; ##p<0.01 vs. vehicle-treated group).
In addition, sciatic nerve injury significantly increased the mRNA level of P450c17 in the lumbar spinal cord dorsal horn at day 5 post-surgery as compared to that of the sham group (Fig. 4B; *p<0.05 vs. Sham), and this increase was significantly inhibited by i.t. administration of L-NIL (60 nmol) during the induction phase of neuropathic pain (from days 0~5 post-surgery) in CCI rats (Fig. 4B; #p<0.05 vs. vehicle-treated group). These results demonstrate that activation of NOS-II increases the protein and mRNA levels of the steroidogenic enzyme P450c17 in the lumbar spinal cord dorsal horn of CCI rats.
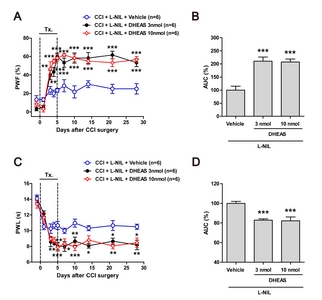
Co-administration of DHEAS restores the CCI-induced development of neuropathic pain that was suppressed by L-NIL administration alone
To determine whether spinal P450c17, a DHEA-synthesizing enzyme, contributes to the NOS-II-induced development of neuropathic pain in CCI rats, dehydroepiandrosterone sulfate (DHEAS) was co-administrated with L-NIL on postoperative days 0~5. Since DHEAS is more stable than DHEA, DHEAS was used in the present study. Repeated daily intrathecal co-administration of DHEAS (3 or 10 nmol) with L-NIL (60 nmol) during the induction phase of neuropathic pain restored the CCI-induced development of mechanical allodynia that was originally inhibited by L-NIL administration alone (Fig. 5A; **p<0.01, ***p<0.001 vs. L-NIL-treated group). In addition, the AUC (%) data analysis showed that the L-NIL-induced analgesic effect on the development of mechanical allodynia was restored by co-administration of DHEAS with L-NIL in neuropathic rats (Fig. 5B; ***p<0.001 vs. L-NIL-treated group).
Moreover, i.t. co-administration of DHEAS (3 or 10 nmol) and L-NIL (60 nmol) during the induction phase of neuropathic pain restored the CCI-induced development of thermal hyperalgesia that was originally inhibited by L-NIL administration alone (Fig. 5C; *p<0.05, **p<0.01, ***p<0.001 vs. L-NIL-treated group). In addition, the AUC (%) data analysis showed that the L-NIL-induced analgesic effect on the development of thermal hyperalgesia was restored by co-administration of DHEAS with L-NIL in neuropathic rats (Fig. 5D; ***p<0.001 vs. L-NIL-treated group).
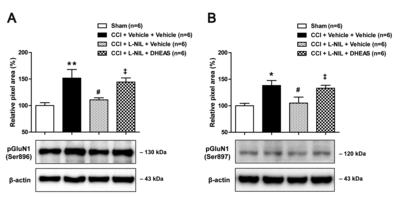
Co-administration of DHEAS restores the CCI-induced increase in PKC- and PKA-dependent GluN1 phosphorylation that were suppressed by L-NIL administration alone
In order to determine whether spinal P450c17 plays a role in the NOS-II-induced increase in the functional potentiation of NMDA receptors, DHEAS was co-administrated with L-NIL on postoperative days 0~5 and the changes in the PKC- and PKA-dependent GluN1 phosphorylation was then examined using Western blot analysis. Repeated daily intrathecal co-administration of DHEAS (10 nmol) with L-NIL (60 nmol) during the induction phase of neuropathic pain restored the CCI-induced increase in the PKC-dependent GluN1 phosphorylation at the Ser896 site (Fig. 6A) and the PKA-dependent GluN1 phosphorylation at the Ser897 site (Fig. 6B) in the lumbar spinal cord dorsal horn at day 5 post-surgery as compared to that of the L-NIL-treated group (Fig. 6; *p<0.05, **p<0.01 vs. Sham, #p<0.05 vs. vehicle-treated group, ‡p<0.05 vs. L-NIL-treated group). These results demonstrate that spinal P450c17, a DHEA-synthesizing enzyme, plays an important role in the NOS-II-induced potentiation of NMDA receptor function, as well as, the development of neuropathic pain in CCI rats.
DISCUSSION
The processing of inflammatory and neuropathic pain is associated with the production of NO in the spinal cord. In the present study, sciatic nerve injury induced a significant increase in the expression of NOS-II in spinal cord microglial cells and in the total NO levels in the lumbar spinal cord dorsal horn at day 5 post-surgery. Inhibition of NOS-II with L-NIL during the induction phase of neuropathic pain (from days 0 to 5 post-surgery) significantly reduced the development of mechanical allodynia and thermal hyperalgesia as well as the CCI-induced increase in PKC- and PKA-dependent pGluN1 expression. In addition, sciatic nerve injury increased the protein and mRNA levels of cytochrome P450c17, a DHEA-synthesizing enzyme, in the lumbar spinal cord dorsal horn at day 5 post-surgery, and this increase was suppressed by repeated i.t. administration of L-NIL. Furthermore, i.t. administration of the sulfated form of DHEA, DHEAS restored the CCI-induced development of mechanical allodynia and thermal hyperalgesia, as well as, increased pGluN1 expression, which were originally attenuated by i.t. administration of L-NIL alone. Collectively these findings suggest that activation of NOS-II increases the expression of spinal P450c17, ultimately contributing to the increase in phosphorylation of NMDA receptor GluN1 subunit and the development of neuropathic pain associated with peripheral nerve injury.
Nitric oxide plays an important role in various physiological and pathological processes in the nervous system. At low concentrations, NO plays a role in neurotransmission and as a neuromodu-lator serving to regulate aspects of nervous system function, while at higher concentrations, NO increases oxidative stress and has a role in the pathogenesis of inflammatory and neuropathic pain [16, 21]. Nitric oxide synthase type II is an inducible NOS that is expressed in glial cells of the central nervous system by a wide variety of pro-inflammatory stimuli [22–24]. In vitro, inflammatory cytokines such as TNFα, IL-1 and IFNγ, which are also upregulated following nerve injury in vivo, can synergize in stimulated glial cells to express other cytotoxic inflammatory mediators such as NOS-II and to release high titers of NO [25, 26]. In several types of nerve injury, mice lacking NOS-II show evidence of a myelinated fiber Wallerian degeneration delay, which is associated with delayed expression of neuropathic pain [27]. In the present study, sciatic nerve injury increased the expression of NOS-II in microglial cells and concomitant NO production in the spinal cord of neuropathic rats. In addition, inhibition of NOS-II during the induction phase of neuropathic pain significantly suppressed the development of neuropathic pain. These results suggest the possibility that microglial NOS-II-induced initiation of a neuroinflammatory process plays a critical role in the pathological changes in the spinal cord under peripheral nerve injury-associated neuropathic pain conditions.
Neurosteroids are locally synthesized in the central nervous system following nerve injuries as independent from steroidogenic organs [28–30]. Among steroidogenic enzymes, cytochrome P450c17 catalyzes the 17-alpha-hydroxylation of pregnenolone and subsequent cleavage of the residual two carbons at C17 leading to the formation of DHEA. DHEA has an effect on the spinal nociceptive signal transmission producing a rapid pronociceptive effect [31], and its sulfate ester DHEAS also plays an important role in the enhancement of NMDA-induced nociception via the activation of spinal sigma-1 receptors, but not through the inhibition of GABAA receptors [8]. In a previous study from our laboratories, P450c17 immunostaining was shown to increase in spinal astrocytes and inhibition of P450c17 significantly decreased the activity of astrocyte sigma-1 receptors in CCI mice [9]. In the present study, inhibition of NOS-II with L-NIL significantly reduced not only the protein expression of P450c17, but also the mRNA levels of P450c17 in the lumbar spinal cord dorsal horn of CCI rats. In addition, DHEAS administration restored the development of neuropathic pain that was originally suppressed by L-NIL administration alone. The current results suggest the possibility that NOS-II could change the neurosteroid-mediated synaptic microenvironment through modulation of astrocyte P450c17 expression at the gene transcription level, ultimately contributing to spinal nociceptive signaling during the induction phase of neuropathic pain in CCI rats.
Regulation of gene transcription in response to extracellular signals is initiated by receptor molecules and mediated by a broad spectrum of intracellular signaling cascades [14]. It has been suggested that diverse ion channels, protein tyrosine kinases, mitogen-activated protein kinases and protein phosphatases can be the targets of NO [14]. In addition, NO has other molecular targets resulting in regulation of diverse gene expression. NO does not only directly influence the activity of transcription factors including nuclear factor (NF)-κB, activating protein (AP)-1 and specificity protein 1 (Sp1), but it also modulates mRNA stability and translation, as well as, the processing of the primary gene products [14]. It has been demonstrated that the transcription of the P450c17 gene is regulated by cAMP-dependent binding of steroidogenic factor-1 (SF-1) to its promoter in the adrenal cortex [32]. Furthermore, the nuclear phosphoprotein SET binds to the specific DNA site of the rat P450c17 gene and transactivates P450c17 in neuronal cells [33]. There is the possibility that NO regulates the expression of the P450c17 gene through the modulation of transcription or post-transcription, however, the detailed mechanisms by which spinal NOS-II/NO signaling regulates the expression of P450c17 need to be further investigated in future studies.
In the present study, we focused our investigation on the NOS-II-induced positive modulation of P450c17 expression in the spinal cord as a potential nociceptive mechanism that underlies the development of neuropathic pain following peripheral nerve injury in rats. By contrast, studies in multiple species have demonstrated primarily an inhibition of steroidogenesis by NO in endocrine tissues [34, 35]. These actions are supported by an in vivo study showing that the stress-induced down-regulation of testicular steroidogenesis is mediated by the intratesticular NO signaling pathway [34]. In the ovary, NO regulation of blood flow may provide a key means of altering steroid formation, while in the adrenal gland, NO may alter steroid production by acting directly on the steroidogenic enzymes themselves or by altering their expression via effects on transcription factor function [35]. Thus, these results suggest that the underlying mechanisms in NO-induced regulation of steroidogenesis may depend on the tissue types and the environment status. Unlike the inhibitory effects that occur with NO in steroidogenic tissues, increased NO signaling in spinal cord appears to contribute to the neurosteroidogenesis induced by P450c17 and nociceptive signaling transmission under pathological conditions such as neuropathic pain. In this regard, it is likely that NO modulates neurosteroidogenesis in the spinal cord in association with nociception in different ways than those associated with other types of NO-related signaling that occur in steroidogenic organs.
In the spinal cord, the activation of NMDA receptors is known to be an essential contributor to the process of ‘central sensitization’, a phenomenon in which nociceptive inputs to the dorsal horn increase the excitability and synaptic efficacy of neurons in spinal pain pathways [36]. It has been suggested that selective inhibition of NOS-II blocks the increased responses of rat dorsal horn neurons, as well as, the development of secondary hyperalgesia following intradermal capsaicin injection representing a spinal contribution to central sensitization [37]. This is consistent with the results of the present study showing that inhibition of NOS-II during the induction phase of neuropathic pain significantly decreases the PKC- and PKA-dependent phosphorylation of the NMDA receptor GluN1 subunit, and importantly this decrease is restored by co-administration of DHEAS with L-NIL. These results suggest a potential role of NOS-II in the early phosphorylation-dependent phase of central sensitization, which is possibly mediated by DHEAS induced by P450c17. Moreover, it has been demonstrated that PKC modulates functional NMDA receptor channel trafficking to the plasma membrane as well as increases the probability of channel openings [38, 39]. Thus, the spinal NOS-II-induced increase in GluN1 phosphorylation could play a critical role in the functional activation of NMDA receptors and the development of central sensitization during the early phase of neuropathic pain.
In conclusion, the present study demonstrates that activation of NOS-II increases the expression of the DHEA-synthesizing enzyme, P450c17 in the lumbar spinal cord dorsal horn during the induction phase of neuropathic pain. Moreover, the sulfated form of DHEA, DHEAS also appears to mediate NOS-II’s effects on PKC- and PKA-dependent phosphorylation of the NMDA receptor GluN1 subunit and the development of CCI-induced neuropathic pain. Collectively these findings suggest that the NOS-II-induced increase in P450c17 plays a critical role in spinal NOS-II-induced central sensitization, and ultimately contributes to the development of neuropathic pain associated with peripheral nerve injury.
Supplementary Information
ACKNOWLEDGEMENTS
This research was supported by the National Research Foundation of Korea (NRF) grant (No. 2017R1A2A2A05001402) funded by the Korean Government, South Korea.
Figures
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Tables
Table 1 Sequences of primers used in RT-PCR and real-time PCR
| Target genes | Forward 5′-3′ | Reverse 5′-3′ |
|---|---|---|
| P450c17 | TTTTGGCCCAAGT-CAAAGAC | CCCTTCTTCACGAG-CACTTC |
| GAPDH | AACTTTGGCATTGTG-GAAGG | ACACATTGGGGGTAG-GAACA |
RT-PCR, reverse transcription polymerase chain reaction.
References
- Baulieu EE (1997) Neurosteroids: of the nervous system, by the nervous system, for the nervous system. Recent Prog Horm Res 52 : 1-32.

- Compagnone NA, Mellon SH (2000) Neurosteroids: biosynthesis and function of these novel neuromodulators. Front Neuroendocrinol 21 : 1-56.

- Corpéchot C, Robel P, Axelson M, Sjövall J, Baulieu EE (1981) Characterization and measurement of dehydroepiandrosterone sulfate in rat brain. Proc Natl Acad Sci U S A 78 : 4704-4707.

- Baulieu EE, Robel P (1998) Dehydroepiandrosterone (DHEA) and dehydroepiandrosterone sulfate (DHEAS) as neuroactive neurosteroids. Proc Natl Acad Sci U S A 95 : 4089-4091.
- Monnet FP, Maurice T (2006) The sigma1 protein as a target for the non-genomic effects of neuro(active)steroids: molecular, physiological, and behavioral aspects. J Pharmacol Sci 100 : 93-118.
- Meyer JH, Gruol DL (1994) Dehydroepiandrosterone sulfate alters synaptic potentials in area CA1 of the hippocampal slice. Brain Res 633 : 253-261.
- Vallée M, Mayo W, Le Moal M (2001) Role of pregnenolone, dehydroepiandrosterone and their sulfate esters on learning and memory in cognitive aging. Brain Res Brain Res Rev 37 : 301-312.
- Yoon SY, Roh DH, Seo HS, Kang SY, Moon JY, Song S, Beitz AJ, Lee JH (2010) An increase in spinal dehydroepiandrosterone sulfate (DHEAS) enhances NMDA-induced pain via phosphorylation of the NR1 subunit in mice: involvement of the sigma-1 receptor. Neuropharmacology 59 : 460-467.
- Choi SR, Roh DH, Yoon SY, Choi HS, Kang SY, Han HJ, Beitz AJ, Lee JH (2019) Spinal cytochrome P450c17 plays a key role in the development of neuropathic mechanical allodynia: involvement of astrocyte sigma-1 receptors. Neuropharmacology 149 : 169-180.
- Alderton WK, Cooper CE, Knowles RG (2001) Nitric oxide synthases: structure, function and inhibition. Biochem J 357 : 593-615.
- Esplugues JV (2002) NO as a signalling molecule in the nervous system. Br J Pharmacol 135 : 1079-1095.
- Cury Y, Picolo G, Gutierrez VP, Ferreira SH (2011) Pain and analgesia: the dual effect of nitric oxide in the nociceptive system. Nitric Oxide 25 : 243-254.
- Meller ST, Gebhart GF (1993) Nitric oxide (NO) and nociceptive processing in the spinal cord. Pain 52 : 127-136.
- Bogdan C (2001) Nitric oxide and the regulation of gene expression. Trends Cell Biol 11 : 66-75.
- Levy D, Höke A, Zochodne DW (1999) Local expression of inducible nitric oxide synthase in an animal model of neuropathic pain. Neurosci Lett 260 : 207-209.
- De Alba J, Clayton NM, Collins SD, Colthup P, Chessell I, Knowles RG (2006) GW274150, a novel and highly selective inhibitor of the inducible isoform of nitric oxide synthase (iNOS), shows analgesic effects in rat models of inflammatory and neuropathic pain. Pain 120 : 170-181.
- Bennett GJ, Xie YK (1988) A peripheral mononeuropathy in rat that produces disorders of pain sensation like those seen in man. Pain 33 : 87-107.
- Choi SR, Roh DH, Yoon SY, Kang SY, Moon JY, Kwon SG, Choi HS, Han HJ, Beitz AJ, Oh SB, Lee JH (2013) Spinal sigma-1 receptors activate NADPH oxidase 2 leading to the induction of pain hypersensitivity in mice and mechanical allodynia in neuropathic rats. Pharmacol Res 74 : 56-67.
- Hargreaves K, Dubner R, Brown F, Flores C, Joris J (1988) A new and sensitive method for measuring thermal nociception in cutaneous hyperalgesia. Pain 32 : 77-88.
- Choi SR, Kwon SG, Choi HS, Han HJ, Beitz AJ, Lee JH (2016) Neuronal NOS activates spinal NADPH oxidase 2 contributing to central sigma-1 receptor-induced pain hypersensitivity in mice. Biol Pharm Bull 39 : 1922-1931.
- Conti A, Miscusi M, Cardali S, Germanò A, Suzuki H, Cuzzocrea S, Tomasello F (2007) Nitric oxide in the injured spinal cord: synthases cross-talk, oxidative stress and inflammation. Brain Res Brain Res Rev 54 : 205-218.
- Saha RN, Pahan K (2006) Regulation of inducible nitric oxide synthase gene in glial cells. Antioxid Redox Signal 8 : 929-947.
- Galea E, Feinstein DL, Reis DJ (1992) Induction of calciumindependent nitric oxide synthase activity in primary rat glial cultures. Proc Natl Acad Sci U S A 89 : 10945-10949.
- Nomura Y, Kitamura Y (1993) Inducible nitric oxide synthase in glial cells. Neurosci Res 18 : 103-107.
- Tran EH, Hardin-Pouzet H, Verge G, Owens T (1997) Astrocytes and microglia express inducible nitric oxide synthase in mice with experimental allergic encephalomyelitis. J Neuroimmunol 74 : 121-129.
- Jeong HK, Ji K, Min K, Joe EH (2013) Brain inflammation and microglia: facts and misconceptions. Exp Neurobiol 22 : 59-67.
- Levy D, Kubes P, Zochodne DW (2001) Delayed peripheral nerve degeneration, regeneration, and pain in mice lacking inducible nitric oxide synthase. J Neuropathol Exp Neurol 60 : 411-421.
- García-Estrada J, Luquín S, Fernández AM, Garcia-Segura LM (1999) Dehydroepiandrosterone, pregnenolone and sex steroids down-regulate reactive astroglia in the male rat brain after a penetrating brain injury. Int J Dev Neurosci 17 : 145- 151.
- Patte-Mensah C, Mensah-Nyagan AG (2008) Peripheral neuropathy and neurosteroid formation in the central nervous system. Brain Res Brain Res Rev 57 : 454-459.
- Kibaly C, Patte-Mensah C, Mensah-Nyagan AG (2005) Molecular and neurochemical evidence for the biosynthesis of dehydroepiandrosterone in the adult rat spinal cord. J Neurochem 93 : 1220-1230.
- Kibaly C, Meyer L, Patte-Mensah C, Mensah-Nyagan AG (2008) Biochemical and functional evidence for the control of pain mechanisms by dehydroepiandrosterone endogenously synthesized in the spinal cord. FASEB J 22 : 93-104.
- Dammer EB, Leon A, Sewer MB (2007) Coregulator exchange and sphingosine-sensitive cooperativity of steroidogenic factor-1, general control nonderepressed 5, p54, and p160 coactivators regulate cyclic adenosine 3',5'-monophosphate- dependent cytochrome P450c17 transcription rate. Mol Endocrinol 21 : 415-438.
- Compagnone NA, Zhang P, Vigne JL, Mellon SH (2000) Novel role for the nuclear phosphoprotein SET in transcriptional activation of P450c17 and initiation of neurosteroidogenesis. Mol Endocrinol 14 : 875-888.
- Kostic TS, Andric SA, Maric D, Stojilkovic SS, Kovacevic R (1999) Involvement of inducible nitric oxide synthase in stress-impaired testicular steroidogenesis. J Endocrinol 163 : 409-416.
- Ducsay CA, Myers DA (2011) eNOS activation and NO function: differential control of steroidogenesis by nitric oxide and its adaptation with hypoxia. J Endocrinol 210 : 259-269.
- Woolf CJ, Mannion RJ (1999) Neuropathic pain: aetiology, symptoms, mechanisms, and management. Lancet 353 : 1959- 1964.
- Wu J, Fang L, Lin Q, Willis WD (2001) Nitric oxide synthase in spinal cord central sensitization following intradermal injection of capsaicin. Pain 94 : 47-58.
- Chen L, Huang LY (1992) Protein kinase C reduces Mg2+ block of NMDA-receptor channels as a mechanism of modulation. Nature 356 : 521-523.
- Lan JY, Skeberdis VA, Jover T, Grooms SY, Lin Y, Araneda RC, Zheng X, Bennett MV, Zukin RS (2001) Protein kinase C modulates NMDA receptor trafficking and gating. Nat Neurosci 4 : 382-390.