Articles

Article Tools
 View Full Text View Full Text |
 Abstract Abstract |
 Article as PDF Article as PDF |
 Print this Article Print this Article |
 Pubmed Pubmed |
 PMC PMC |
 PubReader PubReader |
 Export to Citation Export to Citation |
 Email Alerts Email Alerts |
 Open Access Open Access |
Supplementary
Share this article on :



Stats or Metrics
Article
Original Article
Exp Neurobiol 2019; 28(4): 529-536
Published online August 31, 2019
https://doi.org/10.5607/en.2019.28.4.529
© The Korean Society for Brain and Neural Sciences
Calpain-2 as a Treatment Target in Prenatal Stress-induced Epileptic Spasms in Infant Rats
Hyeok Hee Kwon1,2,4, Chiranjivi Neupane1,5, Juhee Shin1,2, Do Hyeong Gwon1,2, Yuhua Yin1,2, Nara Shin1,2, Hyo Jung Shin1,2,3, Jinpyo Hong2,3, Jin Bong Park1,3,5, YoonYoung Yi4, Dong Woon Kim1,2,3* and Joon Won Kang1,3,4*
1Department of Medical Science, Chungnam National University, Daejeon 35015, 2Department of Anatomy, School of Medicine, Chungnam National University, Daejeon 35015, 3Brain Research Institute, School of Medicine, Chungnam National University, Daejeon 35015, 4Department of Pediatrics, Chungnam National University Hospital, Daejeon 35015, 5Department of Physiology, School of Medicine, Chungnam National University, Daejeon 35015, Korea
Correspondence to: *To whom correspondence should be addressed.
Dong Woon Kim, TEL: 82-42-580-8209, FAX: 82-42-586-4800
e-mail: visnu528@cnu.ac.kr
Joon Won Kang, TEL: 82-42-280-8244, FAX: 82-42-255-3158
e-mail: childlove@cnu.ac.kr
This is an Open Access article distributed under the terms of the Creative Commons Attribution Non-Commercial License(http://creativecommons.org/licenses/by-nc/4.0) which permits unrestricted non-commercial use, distribution, andreproduction in any medium, provided the original work is properly cited.
Abstract
Stress can induce a serious epileptic encephalopathy that occurs during early infancy. Recent studies have revealed that prenatal stress exposure is a risk factor for the development of infantile spasms. Our previous work demonstrates that prenatal stress with betamethasone-induced alterations to the expression of the K+/Cl− co-transporter (KCC2) in gamma-aminobutyric acid (GABA) interneurons lowers the seizure threshold in exposed animals. Here, we further investigated the mechanisms involved in this KCC2 dysfunction and explored possible treatment options. We stressed Sprague-Dawley rats prenatally and further treated dams with betamethasone on gestational day 15, which increases seizure susceptibility and NMDA (N-Methyl-D-aspartate)-triggered spasms on postnatal day 15. In this animal model, first, we evaluated baseline calpain activity. Second, we examined the cleavage and dephosphorylation of KCC2. Finally, we checked the effect of a calpain inhibitor on seizure occurrence. The phosphorylated-N-methyl-D-aspartate Receptor 2B (NR2B):non-phosphorylated NR2B ratio was found to be higher in the cortex of the prenatally stressed beta-methasone model. We further found that the betamethasone model exhibited increased phosphorylation of calpain-2 and decreased phosphorylation of KCC2 and Glutamic acid decarboxylase 67 (GAD67). After using a calpain inhibitor in prenatal-stress rats, the seizure frequency decreased, while latency increased. GABAergic depolarization was further normalized in prenatal-stress rats treated with the calpain inhibitor. Our study suggests that calpain-dependent cleavage and dephosphorylation of KCC2 decreased the seizure threshold of rats under prenatal stress. Calpain-2 functions might, thus, be targeted in the future for the development of treatments for epileptic spasms.
Graphical Abstract
Keywords: Epilepsy, Calpain, KCC2, NMDA, Glutamate decarboxylase 67, K+/Cl- co-transporter
INTRODUCTION
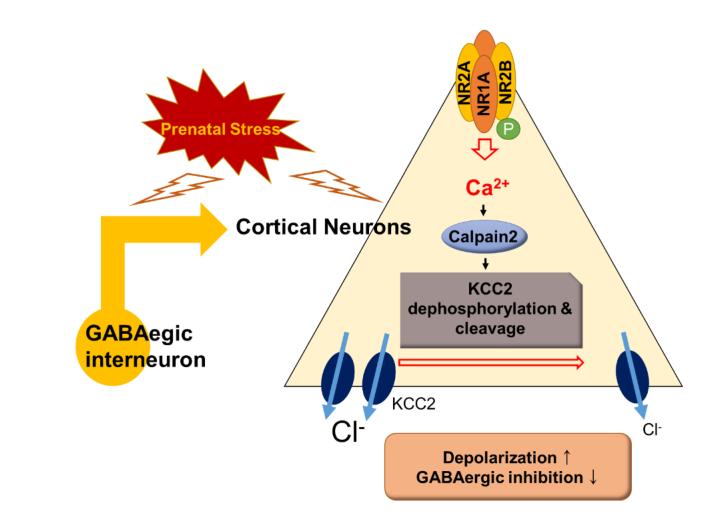
Epilepsy is a common neurodegenerative disorder, remarkably in childhood, and is known to occur in 0.5 to 1% of patients [1]. A significant factor affecting epileptic spasms and epilepsy is stress [2, 3]. Prenatal stress can affect neuronal development and physiological dysregulation [4] and can alter the secretion of glucocorticoids in the hypothalamic–pituitary–adrenocortical (HPA) axis [5]. We have previously suggested a possible mechanism by which pre-natal stress and associated dysfunction of gamma-aminobutyric acid (GABA) interneurons via altered expression of the K+/Cl− co-transporter (KCC2) can contribute to enhanced epileptic spasms in a prenatally stressed betamethasone model [6].
Increased calpain expression has been reported in seizures and epilepsy [7, 8]. Calpain is a calcium-dependent, non-lysosomal cysteine protease that modifies the structure and regulates the activity of protein targets by limited proteolysis [9]. Calpain activation requires an elevated cellular Ca2+ concentration via the activity of N-Methyl-D-aspartate (NMDA) receptors [10]. Extrasynaptic NMDA receptors activate calpain, leading to cell death [11, 12]. KCC2 is also known as a substrate of calpain [11].
KCC2 plays critical roles in the generation and maintenance of synaptic plasticity [13] and is involved in neuronal chloride regulation [14]. In immature neurons, it maintains a low intracellular Cl− concentration that forms the basis for the hyperpolarization of the GABAA receptor [15]. However, low expression of KCC2 induces high intracellular Cl− concentration and depolarization of the GABAA receptor response during development [16].
According to recent reports, KCC2 is related to febrile seizure, idiopathic generalized epilepsy, and infantile spasms [6, 17]. It has been demonstrated to lead to deficits in neuronal Cl− extrusion capacity. A series of experiments indicated that increased neuronal activity leads to calpain-mediated cleavage of KCC2, resulting in a relief in diffusion constraints and subsequent dispersal of KCC2 clusters [13]. Furthermore, Chamma et al. [13] used a phosphomimetic KCC2 S940D mutant that was found to prevent the activity-induced dephosphorylation-dependent endocytosis and degradation of KCC2.
Here, we use rats primed with the synthetic corticosteroid betamethasone to investigate the relationships between epileptic spasm-associated molecular markers, including calpain activity, total KCC2 protein levels, phosphorylated KCC2 levels, and KCC2 cleavage. By using electrophysiological methods, we further demonstrated that a calpain inhibitor may serve as a target for the treatment of epileptic spasms.
MATERIALS AND METHODS
Animals
Pregnant Sprague-Dawley rats purchased from Samtako Bio Korea (Osan, Korea) were housed at 23°C under a controlled 12 h:12 h light:dark cycle with light on at 08:00. Food and water were accessible
MDL-28170, a calpain-2 inhibitor was used [11]. Rats received intraperitoneal (i.p.) injection of MDL-28170 (20 mg/kg) or DMSO vehicle twice daily (at 09:00 and 19:00), from postnatal day 7 to 15.
All studies conducted herein were approved by our institutional animal use committee. Furthermore, the authors considered all guidelines outlined in the ARRIVE guidelines and Basel declaration, including the 3R concept, when planning these experiments.
Antibodies and reagents
All commercial antibodies and reagents were purchased from the following sources: NR2A (1:500; AB1555P), NR2B (1:1000; #06-600), KCC2 (1:1000, #07-432), and GAD67 (1:1000, MAB5406) were purchased from Millipore (Billerica, MA, USA). NR2B-phospho S1303 (1:1000; ab81271) and KCC2-NTD (1:1000, ab107452) were purchased from Abcam (Cambridge, MA, USA). Calpain-1 (1:1000, #2556S) and calpain-2 (1:1000; #2539S) were purchased from Cell Signaling Technology (Danvers, MA, USA). KCC2-phospho S940 (1:1000; p1551-940) was purchased from Phosphosolutions (Aurora, CO, USA). β-actin (1:10000, A5316; Sigma-Aldrich, St. Louis, MO, USA) was used as a loading control. Goat anti-rabbit IgG-HRP (1:5000, #LF-SA8002) and goat anti-mouse IgG-HRP (1:5000, #LF-SA8001) were purchased from AbFron-tier Co., Ltd. (Seoul, Korea). N-Methyl-D-aspartic acid (NMDA) (M3262) and MDL-27180 (M6690) were purchased from Sigma-Aldrich.
Western blot analysis
The whole cortex from each of the P15 pups (n=8) of each group was dissected and homogenized in PRO-PREPTM Protein Extraction Solution (iNtRON Biotechnology Inc., Korea). After centrifugation (13,000 rpm, 15min and 4°C), protein concentrations in the supernatants were determined using Micro BCA protein assay kit (Pierce Chemical, TX, USA) with bovine serum albumin used as the standard. Aliquots containing 30 μg of protein were resolved by 8% or 10% SDS polyacrylamide gel electrophoresis and transferred onto nitrocellulose membranes. For immunoblotting, membranes were incubated in 5% skimmed milk prepared in TBS-T (0.1% Tween-20 in TBS) for 1 h to block non-specific binding sites, followed by incubation with respective primary antibodies. Membranes were then washed three times for 10 min each in TBS-T, followed by incubation for 1 h with respective horseradish peroxidase-labeled secondary antibodies (AbFrontier Co., Ltd.) of 1:5000 dilution prepared in ProNATM 5X Phospho-BLOCK Solution (TransLab, Daejeon, Korea). After three further washes, immuno-labeled proteins were detected using a SuperSignal enhanced chemiluminescence kit (Pierce Chemical) and ChemiDocTM Touch Imaging System (Bio-Rad Laboratories, Inc., Hercules, CA, USA). Three individual samples from the whole cortex at P15 were used for western blot analysis.
Calpain activity assay
Total nine betamethasone rats and nine control rats were used. The Calpain–Glo Protease Assay (Promega, CA, USA) was used to check the calpain activity in whole cortex lysate (5 μg) according to manufacturer’s instructions. Protein concentrations were measured using BCA assay kit (Pierce Chemical) with BSA as the standard. Calpain activity was measured by luminoskan ascent luminometer (Thermo scientific).
Electrophysiology and data analysis
Electrophysiological recordings were obtained from acutely prepared coronal brain slices containing cortical neurons. P14 rats were anesthetized with avertin (250 mg/kg, i.p.) (Sigma Aldrich) before decapitation. Brains were quickly separated and coronal hippocampal slices (300 μM) were prepared using Vibrotome (Leica VT 1200S; Leica, Germany). Sectioned slices were incubated in artificial cerebrospinal fluid (aCSF) at 34°C for 1 h before use. aCSF consists of NaCl (126 mM), NaHCO3 (26 mM), KCl (5 mM), NaH2PO4 (1.2 mM), D-glucose (10 mM), CaCl2 (2.4 mM), and MgCl2 (1.2 mM), pH 7.3~7.4 (300~315 mOsm/kg). Slices were continuously bubbled with mixture of 95% O2 and 5% CO2 from the beginning to the end of the recording. Sectioned slices were transferred to the recording chamber continuously perfused with aCSF with flow rate of 3~4 ml/min at 32~34°C. For perforated patch-clamp recording, gramicidin (Sigma Aldrich) was first dissolved in dimethyl sulfoxide (DMSO) (3.5 mg/ml) and then diluted to 3.5 μg/ml. Patch pipettes were dipped in to the diluted gramicidin and filled with internal solution containing K-gluconate (130 mM), KCl (10 mM), HEPES (10 mM), EGTA (10 mM), Mg2+ ATP (5 mM), and MgCl2 (0.9 mM), pH 7.3~7.4. Stable holding level obtained after 5 to 10 min of gigaseal formation corresponds to the resting membrane potential. Series resistance was monitored from the beginning to the end of the experiments. Currents were acquired using Axopatch 200B (Axon instruments, Foster City, CA, USA), filtered at 1 kHz, and digitized at 10 kHz (Digidata 1400A, pClamp 10.2 software). Quantitative data are expressed as mean±SEM. Student’s
Statistical analysis
We used the NIH image analysis software (ImageJ) to assess immunoblots quantitatively using densitometry detection methods. The statistical analysis of immunoblot was performed using the Mann-Whitney U-test. From animal experiments, resulting quantitative data across the groups were analyzed using analysis of variance (ANOVA). The Newman-Keuls method was used for post-hoc analyses. A p-value of less than 0.05 was considered as statistically significant. All statistical analyses were conducted using the Prism 5.0 software (GraphPad, San Diego, CA, USA).
RESULTS
Epileptic spasms were triggered by NMDA in the betamethasone model
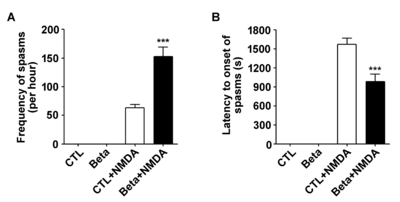
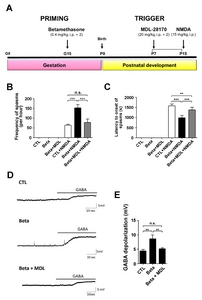
We used a rat model of G15 exposure to betamethasone to investigate NMDA-induced spasms [6, 18, 19]. To determine whether there is a prenatal stress effect on the susceptibility to NMDA-induced spasms, we measured the frequency of spasms and latency to spasms in NMDA-treated P15 rat offspring and in age-matched controls. The limitation of our model is that it does not induce spontaneous spasms. The frequency of spasms was visibly increased in infant rats exposed to betamethasone (mean number in controls±SD: 63.17±13.42, p<0.0001, mean number in betamethasone-exposed animals±SD: 152.83±40.59, p<0.0001) (Fig. 1A). In addition, the latency to the onset of spasms was decreased in the betamethasone-exposed animals [mean latency (sec) in controls±SD: 1574.67±234.65, mean latency in betamethasone-exposed animals±SD: 986.17±289.83, p<0.0001] (Fig. 1B).
Increased phosphorylation of NMDA receptor 2B and calpain may suggest a role of calpain-2 in the increased susceptibility of spasms
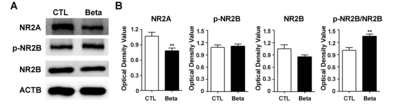
To measure alterations to NMDA receptors in the whole cortex of betamethasone-exposed rats, we measured levels of NR2A, NR2B, and phosphorylated-NR2B (S1303) proteins (Fig. 2). Increase in NR2A and NR2B ratio results in maturation and changes of NMDA receptor properties [20]. Levels of NR2A were significantly decreased (p<0.01, p=0.0022) with betamethasone. The phosphorylated-NR2B:non-phosphorylated NR2B ratio in the cortex was significantly increased (p<0.01, p=0.0022) with beta-methasone.
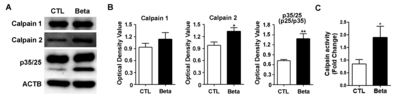
In accordance with the increased Ca2+ that might be expected with NMDA receptor alterations, calpain-2 levels were also significantly increased in the whole cortex upon betamethasone treatment (Fig. 3A). However, the level of calpain-1 did not increase (Fig. 3B). We further examined expression of p35/25 (Fig. 3B). Since cleavage of p35 to p25 induces neurotoxicity, we found significantly increased levels of p25 in betamethasone model rats (p<0.01, p=0.0022). Increased calpain-2 in this betamethasone model indicates increased susceptibility to seizures via calpain-2 (p<0.05, p=0.0260) (Fig. 3B). The calpain enzyme activity was increased in the betamethasone model rats (p<0.05, p=0.0260) (Fig. 3B).
Decreased KCC2 by calpain-2 in the betamethasone model rats
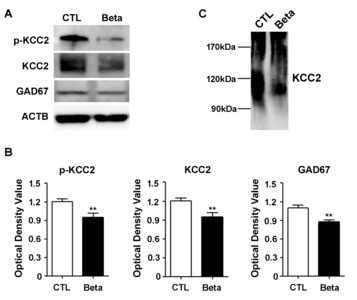
KCC2 is a known substrate of calpain [11]. Given this, we investigated the cleavage of KCC2 by calpain. We measured the level of phospho-KCC2 (p<0.01, p=0.0087), total KCC2 (p<0.01, p=0.0087), and GAD67 (p<0.01, p=0.0022) protein expression and found that all protein levels were decreased in the whole cortex of betamethasone model rats (Fig. 4A and 4B). In addition, we found a decrease in protein size due to cleavage of KCC2 (Fig. 4C).
Inhibition of calpain in the betamethasone model rats
To investigate the effect of calpain-2 inactivation, we applied MDL-28170 (MDL) to our betamethasone model rats [8, 11]. We measured the latency (sec) to the onset of spasms after co-treatment with MDL in the P15 betamethasone model rats and observed an increased latency to the onset of spasms after this co-treatment (Fig. 5A and 5B). The Frequency of spasms was also decreased in offspring prenatally exposed to betamethasone and betamethasone+MDL compared to that of control group (Fig. 5C).
In addition, reduced KCC2 protein level and increased ratio of p35/25 were recovered in MDL-28170 administration group (Supplementary Fig. 1).
Furthermore, gramicidin perforated-patch clamp recordings were performed in cortical neurons from control, betamethasone, and betamethasone+MDL model rats (Fig. 5D). The mean resting membrane potentials (±SEM) in control, betamethasone, and betamethasone+MDL models were −53.63±1.69 mV, −53.65±2.36 mV, and −57.06±2.01 mV, respectively. Bath application of GABA (100 μM) caused significantly larger depolarization of membrane potential in the betamethasone (8.66±1.32 mV, n=4) model compared to that in control (4.40±0.53 mV, n=7, p=0.001). In addition, MDL co-treated with betamethasone reversed the membrane depolarization (5.15±0.36 mV, n=8, p=0.006 compare to beta-methasone) to almost the control level. These data suggested that infantile spasms induced due to GABA-induced membrane depolarization might be prevented by using an inhibitor of calpain-2 (MDL-28170).
DISCUSSION
Infantile spasms is an epileptic encephalopathy that occurs in infancy and affects the development of children [21]. While common epilepsy treatment methods are used for it, it often remains intractable [22]. Epilepsy has several known etiologies, but the association between prenatal stress and epilepsy remains poorly understood. Our previous work demonstrated that prenatal stress causes a decrease in KCC2 function in GABAergic interneurons and consequently affects seizure susceptibility in animals [6]. In the present study, we investigated the causes of KCC2 dysfunction and studied treatment methods for targeting this dysfunction in the context of seizure treatment.
In the brain, calpain over-activation is generally found after excitotoxic conditions including physical trauma or a neurochemical challenge [23]. Calpain-mediated cleavage of proteins is required for proper brain function and is a main component of the cellular damage cascade that often follows excitotoxic events [23, 24]. Calpain activation requires elevated cellular Ca2+ concentration via altered NMDA receptor transport [10]. The extrasynaptic component of the NMDA receptor is composed of a NR1/NR2B subunit. NR2A is highly expressed in synaptic NMDA receptors and NR2B is expressed in extrasynaptic NMDA receptors [25]. The NMDA receptor is also known to be present in GABAergic interneurons and alterations to its activity in these neurons has been associated with brain abnormalities and psychopathology [26]. For example, research on schizophrenia has demonstrated that a functional abnormality in the NMDA receptor affects the function of GAB-Aergic interneurons, possibly leading to disease. Decreased NR2A expression can reduce GAD67 expression and, thus, drive the abnormal development of GABAergic interneurons [27]. Here, we investigated NR2A, NR2B, phospho-NR2B, and GAD67 expression profiles to more closely evaluate the disruption of interneuron systems. In the present study, a reduction of NR2A and GAD67 was also found in prenatally stressed rats, which may in turn affect the function of GABAergic interneurons more broadly. The phosphorylated NR2B:NR2B ratio and calpain-2 level and activity were increased in prenatal stress animals, which we infer is due to elevated cellular Ca2+ concentrations.
Alterations in Ca2+ homeostasis lead to persistent and pathologic over-activation of calpain in a number of neurodegenerative diseases [28]. For example, calpain activation has been found to be excessive in neurological diseases including epilepsy [29–31]. In particular, calpain-specific cleavage of Cyclin-dependent kinase 5 (Cdk5), a proline-directed serine/threonine cyclin-dependent kinase, -p35 to p25 has been implicated in the neurological damage seen in many neurological disorders because they are associated with neuronal migration, synaptic activity, and cell survival and development [32, 33]. Calpains have long been implicated in neuronal cell death and injury, including that resulting from excessive excitotoxicity. For example, Putkonen et al. reported that kainic-acid (KA) produced a dose-dependent increase in intracellular Ca2+ concentration and calpain activity, followed by the induction of Cdk5 phosphorylation and Cdk-p35 cleavage, which are believed to be involved in KA-mediated degeneration of glutamatergic synapses in the rat hippocampus [34]. Echoing these results, in the prenatal stress model rats examined here, calpain-2 levels were increased, as was the cleavage of p35 to p25.
KCC2 plays a critical role in the generation and maintenance of synaptic plasticity [7, 35]. For example, it is critically involved in neuronal chloride regulation [36]. In immature neurons, KCC2 maintains low intracellular Cl− concentration, forming the basis for the hyperpolarizing effect of GABAA receptors [8]. However, during development, KCC2 regulates low intracellular Cl− concentration and leads to a depolarizing GABAA receptor response [37]. According to the recent reports, KCC2 expression is related to febrile seizure, idiopathic generalized epilepsy, and infantile spasms [8, 38, 39]. Alterations to KCC2 have further been demonstrated to lead to deficits in neuronal Cl− extrusion capacity [6]. Increased neuronal activity leads to calpain-mediated cleavage of KCC2, resulting in a relief in protein diffusion constraints and subsequent dispersal of KCC2 clusters [11, 40]. Furthermore, Chamma et al. [13] used a phosphomimetic KCC2 S940D mutant that was found to prevent the activity-induced dephosphorylation-dependent endocytosis and degradation of KCC2.
Our previous work found that KCC2 activity is reduced under prenatal stress. As a result, GABAergic neurons show characteristics of excitation, which lead to increased seizure susceptibility. In this study, we found that the activity of KCC2 is decreased by an increase in calpain. We further found that dephosphorylation and cleavage of KCC2 were increased. In other words, GABAergic neurons were rendered excitatory by the effects of calpain-2. This indicates that calpain may be relevant to potential treatment modalities for seizure.
Many calpain inhibitors have been shown to induce anti-epileptic effects [41]. We assumed here that MDL-28170 could also affect calpain activity and, therefore, investigated the cleavage of the representative calpain substrate protein in prenatal-stress rats. We found that the prenatal betamethasone exposure-enhanced Cdk5-p35 cleavage to p25 was significantly blocked by i.p. administration of MDL-28170. Total number of seizures was decreased and latency to spasms was increased upon MDL-28170 administration. Based on these results, we suggest that a calpain-2 inhibitor produces anti-epileptic effects via calpain inhibition.
One of the limitations of this study is that this betamethasone model does not exhibit spontaneous seizures. However, in other studies that used a similar model, results from EEG and drug response tests were found to be aligned with those for infantile spasms endophenotypes in humans [39, 42]. Another potential limitation of the present study is that it is difficult to quantify stress accurately. Further studies, using more precise measures of stress levels in animals and humans are, thus, needed to better understand the role of prenatal stress in driving offspring seizure risk.
Supplementary Information
ACKNOWLEDGEMENTS
This research was supported by the Basic Science Research Program through the National Research Foundation of Korea (NRF), funded by the Ministry of Science, ICT, and Future Planning (2015R1C1A1A01052351, 2016R1A2B4009409). This work was also supported by Chungnam National University Hospital Research Fund, 2017 (2017-CF-024).
Figures
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
References
- Sirven JI (2015) Epilepsy: a spectrum disorder. Cold Spring Harb Perspect Med 5 : a022848.



- Maguire J, Salpekar JA (2013) Stress, seizures, and hypothalamic- pituitary-adrenal axis targets for the treatment of epilepsy. Epilepsy Behav 26 : 352-362.
- Zhu X, Dong J, Xia Z, Zhang A, Chao J, Yao H (2017) Repeated restraint stress increases seizure susceptibility by activation of hippocampal endoplasmic reticulum stress. Neurochem Int 110 : 25-37.
- Maur DG, Romero CB, Burdet B, Palumbo ML, Zorrilla- Zubilete MA (2012) Prenatal stress induces alterations in cerebellar nitric oxide that are correlated with deficits in spatial memory in rat's offspring. Neurochem Int 61 : 1294-1301.
- McGowan PO, Matthews SG (2018) Prenatal stress, glucocorticoids, and developmental programming of the stress response. Endocrinology 159 : 69-82.
- Baek H, Yi MH, Pandit S, Park JB, Kwon HH, Zhang E, Kim S, Shin N, Kim E, Lee YH, Kim Y, Kim DW, Kang JW (2016) Altered expression of KCC2 in GABAergic interneuron contributes prenatal stress-induced epileptic spasms in infant rat. Neurochem Int 97 : 57-64.
- Feng ZH, Hao J, Ye L, Dayao C, Yan N, Yan Y, Chu L, Shi FD (2011) Overexpression of μ-calpain in the anterior temporal neocortex of patients with intractable epilepsy correlates with clinicopathological characteristics. Seizure 20 : 395-401.
- Puskarjov M, Ahmad F, Khirug S, Sivakumaran S, Kaila K, Blaesse P (2015) BDNF is required for seizure-induced but not developmental up-regulation of KCC2 in the neonatal hippocampus. Neuropharmacology 88 : 103-109.
- Lee MS, Kwon YT, Li M, Peng J, Friedlander RM, Tsai LH (2000) Neurotoxicity induces cleavage of p35 to p25 by calpain. Nature 405 : 360-364.
- Yu L, Rostamiani K, Hsu YT, Wang Y, Bi X, Baudry M (2011) Calpain-mediated regulation of stargazin in adult rat brain. Neuroscience 178 : 13-20.
- Puskarjov M, Ahmad F, Kaila K, Blaesse P (2012) Activity-dependent cleavage of the K-Cl cotransporter KCC2 mediated by calcium-activated protease calpain. J Neurosci 32 : 11356-11364.
- Zadran S, Bi X, Baudry M (2010) Regulation of calpain-2 in neurons: implications for synaptic plasticity. Mol Neurobiol 42 : 143-150.
- Chamma I, Heubl M, Chevy Q, Renner M, Moutkine I, Eugène E, Poncer JC, Lévi S (2013) Activity-dependent regulation of the K/Cl transporter KCC2 membrane diffusion, clustering, and function in hippocampal neurons. J Neurosci 33 : 15488-15503.
- Wright R, Newey SE, Ilie A, Wefelmeyer W, Raimondo JV, Ginham R, Mcllhinney RA, Akerman CJ (2017) Neuronal chloride regulation via KCC2 is modulated through a GABAB receptor protein complex. J Neurosci 37 : 5447-5462.
- Lee H, Chen CX, Liu YJ, Aizenman E, Kandler K (2005) KCC2 expression in immature rat cortical neurons is sufficient to switch the polarity of GABA responses. Eur J Neurosci 21 : 2593-2599.
- Lee HH, Deeb TZ, Walker JA, Davies PA, Moss SJ (2011) NMDA receptor activity downregulates KCC2 resulting in depolarizing GABAA receptor-mediated currents. Nat Neurosci 14 : 736-743.
- Saitsu H, Watanabe M, Akita T, Ohba C, Sugai K, Ong WP, Shiraishi H, Yuasa S, Matsumoto H, Beng KT, Saitoh S, Miyatake S, Nakashima M, Miyake N, Kato M, Fukuda A, Matsumoto N (2016) Impaired neuronal KCC2 function by biallelic SLC12A5 mutations in migrating focal seizures and severe developmental delay. Sci Rep 6 : 30072.
- Chachua T, Yum MS, Velíšková J, Velíšek L (2011) Validation of the rat model of cryptogenic infantile spasms. Epilepsia 52 : 1666-1677.
- Yum MS, Chachua T, Velíšková J, Velíšek L (2012) Prenatal stress promotes development of spasms in infant rats. Epilepsia 53 : e46-e49.
- Liu XB, Murray KD, Jones EG (2004) Switching of NMDA receptor 2A and 2B subunits at thalamic and cortical synapses during early postnatal development. J Neurosci 24 : 8885- 8895.
- Zhu G, Liu Y, Wang Y, Bi X, Baudry M (2015) Different patterns of electrical activity lead to long-term potentiation by activating different intracellular pathways. J Neurosci 35 : 621- 633.
- Dulac O (2001) What is West syndrome? Brain Dev 23 : 447-452.
- Liu J, Liu MC, Wang KK (2008) Calpain in the CNS: from synaptic function to neurotoxicity. Sci Signal 1 : re1.
- Curcio M, Salazar IL, Mele M, Canzoniero LM, Duarte CB (2016) Calpains and neuronal damage in the ischemic brain: the swiss knife in synaptic injury. Prog Neurobiol 143 : 1-35.
- Benarroch EE (2011) NMDA receptors: recent insights and clinical correlations. Neurology 76 : 1750-1757.
- Moreau AW, Kullmann DM (2013) NMDA receptor-dependent function and plasticity in inhibitory circuits. Neuropharmacology 74 : 23-31.
- Cohen SM, Tsien RW, Goff DC, Halassa MM (2015) The impact of NMDA receptor hypofunction on GABAergic neurons in the pathophysiology of schizophrenia. Schizophr Res 167 : 98-107.
- Araújo IM, Gil JM, Carreira BP, Mohapel P, Petersen A, Pinheiro PS, Soulet D, Bahr BA, Brundin P, Carvalho CM (2008) Calpain activation is involved in early caspase-independent neurodegeneration in the hippocampus following status epilepticus. J Neurochem 105 : 666-676.
- Lau A, Tymianski M (2010) Glutamate receptors, neurotoxicity and neurodegeneration. Pflugers Arch 460 : 525-542.
- McGeer EG, Zhu SG (1990) Lamotrigine protects against kainate but not ibotenate lesions in rat striatum. Neurosci Lett 112 : 348-351.
- Lam PM, Carlsen J, González MI (2017) A calpain inhibitor ameliorates seizure burden in an experimental model of temporal lobe epilepsy. Neurobiol Dis 102 : 1-10.
- Kanungo J, Zheng YL, Amin ND, Pant HC (2009) Targeting Cdk5 activity in neuronal degeneration and regeneration. Cell Mol Neurobiol 29 : 1073-1080.
- Nam HY, Na EJ, Lee E, Kwon Y, Kim HJ (2017) Antiepileptic and neuroprotective effects of oleamide in rat striatum on kainate-induced behavioral seizure and excitotoxic damage via calpain inhibition. Front Pharmacol 8 : 817.
- Cheung ZH, Ip NY (2012) Cdk5: a multifaceted kinase in neurodegenerative diseases. Trends Cell Biol 22 : 169-175.
- Kaila K, Price TJ, Payne JA, Puskarjov M, Voipio J (2014) Cation-chloride cotransporters in neuronal development, plasticity and disease. Nat Rev Neurosci 15 : 637-654.
- Ben-Ari Y, Gaiarsa JL, Tyzio R, Khazipov R (2007) GABA: a pioneer transmitter that excites immature neurons and generates primitive oscillations. Physiol Rev 87 : 1215-1284.
- Viitanen T, Ruusuvuori E, Kaila K, Voipio J (2010) The K+-Cl- cotransporter KCC2 promotes GABAergic excitation in the mature rat hippocampus. J Physiol 588 : 1527-1540.
- Jantzie LL, Getsy PM, Firl DJ, Wilson CG, Miller RH, Robinson S (2014) Erythropoietin attenuates loss of potassium chloride co-transporters following prenatal brain injury. Mol Cell Neurosci 61 : 152-162.
- Kahle KT, Merner ND, Friedel P, Silayeva L, Liang B, Khanna A, Shang Y, Lachance-Touchette P, Bourassa C, Levert A, Dion PA, Walcott B, Spiegelman D, Dionne-Laporte A, Hodgkinson A, Awadalla P, Nikbakht H, Majewski J, Cossette P, Deeb TZ, Moss SJ, Medina I, Rouleau GA (2014) Genetically encoded impairment of neuronal KCC2 cotransporter function in human idiopathic generalized epilepsy. EMBO Rep 15 : 766-774.
- Puskarjov M, Seja P, Heron SE, Williams TC, Ahmad F, Iona X, Oliver KL, Grinton BE, Vutskits L, Scheffer IE, Petrou S, Blaesse P, Dibbens LM, Berkovic SF, Kaila K (2014) A variant of KCC2 from patients with febrile seizures impairs neuronal Cl- extrusion and dendritic spine formation. EMBO Rep 15 : 723-729.
- Putkonen N, Kukkonen JP, Mudo G, Putula J, Belluardo N, Lindholm D, Korhonen L (2011) Involvement of cyclindependent kinase-5 in the kainic acid-mediated degeneration of glutamatergic synapses in the rat hippocampus. Eur J Neurosci 34 : 1212-1221.
- Velíšek L, Shang E, Velíšková J, Chachua T, Macchiarulo S, Maglakelidze G, Wolgemuth DJ, Greenberg DA (2011) GABAergic neuron deficit as an idiopathic generalized epilepsy mechanism: the role of BRD2 haploinsufficiency in juvenile myoclonic epilepsy. PLoS One 6 : e23656.