Articles

Article Tools
 View Full Text View Full Text |
 Abstract Abstract |
 Article as PDF Article as PDF |
 Print this Article Print this Article |
 Pubmed Pubmed |
 PMC PMC |
 PubReader PubReader |
 Export to Citation Export to Citation |
 Email Alerts Email Alerts |
 Open Access Open Access |
Share this article on :



Stats or Metrics
Article
Original Article
Exp Neurobiol 2019; 28(5): 578-592
Published online October 31, 2019
https://doi.org/10.5607/en.2019.28.5.578
© The Korean Society for Brain and Neural Sciences
Modulation of Dopaminergic Neuronal Excitability by Zinc through the Regulation of Calcium-related Channels
Jihyun Noh1* and Jun-mo Chung2
1Department of Science Education, Dankook University, Yongin 16890, 2Department of Brain and Cognitive Sciences, Ewha Womans University, Seoul 03760, Korea
Correspondence to: *To whom correspondence should be addressed.
TEL: 82-31-8005-3842, FAX: 82-31-8021-7231
e-mail: jihyun2@dankook.ac.kr
This is an Open Access article distributed under the terms of the Creative Commons Attribution Non-Commercial License(http://creativecommons.org/licenses/by-nc/4.0) which permits unrestricted non-commercial use, distribution, andreproduction in any medium, provided the original work is properly cited.
Abstract
Depending on the intracellular buffering of calcium by chelation, zinc has the following two apparent effects on neuronal excitability: enhancement or reduction. Zinc increased tonic activity in the depolarized state when neurons were intracellularly dialyzed with EGTA but attenuated the neuronal activity when BAPTA was used as an intracellular calcium buffer. This suggests that neuronal excitability can be modulated by zinc, depending on the internal calcium buffering capacity. In this study, we elucidated the mechanisms of zinc-mediated alterations in neuronal excitability and determined the effect of calcium-related channels on zinc-mediated alterations in excitability. The zinc-induced augmentation of firing activity was mediated via the inhibition of small-conductance calcium-activated potassium (SK) channels with not only the contribution of voltage-gated L-type calcium channels (VGCCs) and ryanodine receptors (RyRs), but also through the activation of VGCCs via melastatin-like transient receptor potential channels. We suggest that zinc modulates the dopaminergic neuronal activity by regulating not only SK channels as calcium sensors, but also VGCCs or RyRs as calcium sources. Our results suggest that the cytosolic calcium-buffering capacity can tightly regulate zinc-induced neuronal firing patterns and that local calcium-signaling domains can determine the physiological and pathological state of synaptic activity in the dopaminergic system.
Graphical Abstract
Keywords: Calcium-activated non-selective cation, Dopamine system, Electrophysiology, Patch clamp recording, Spike frequency, Rat
INTRODUCTION
Zinc ion (Zn2+) is the most abundant transition metal ion in biological organisms, with mammalian cells containing an estimated 100~500 μM of total Zn2+ [1, 2]. Zn2+ is highly enriched at many, but not all, glutamatergic nerve terminals. In the brain, Zn2+ is released upon neuronal activity and is crucial for the control of physiological and pathophysiological brain functions. Zn2+ enables the modulation of neuronal excitability in the substantia nigra pars compacta (SNc) in two ways—excitation or inhibition—by altering the gating properties of voltage-gated transient A-type K+ channels [3]. Noh et al. proposed that Zn2+ increases the bursting activity of SNc neurons in a hyperpolarized state through the reduction of the transient outward K+ current (IA) and decreases its tonic activity in depolarized states when IA is increased. However, this proposal does not support previous observations that in depolarized states, Zn2+ enhances the firing activity of dopaminergic neurons that are acutely isolated from the rat SNc [4]. These contradictory Zn2+-mediated effects on excitability in SNc neurons at depolarized states were the result of investigations employing different intracellular Ca2+ chelators, namely, ethylene glycol tetraacetic acid (EGTA), and 1,2-bis(o-aminophenoxy)ethane-N,N,N’,N’-tetraacetic acid (BAPTA). Therefore, it is important to determine whether the Zn2+-elicited alteration of firing activity is dependent on the Ca2+-buffering capacity of neurons, and on the cytosolic Ca2+-dependent signaling processes.
In central nervous system neurons and many other cell types, transient elevations in intracellular Ca2+ trigger a wide variety of Ca2+-dependent signaling events and processes including the regulation of firing activities and patterns. The Ca2+ signals are precisely restricted to local spatiotemporal domains, which are generated by a variety of different Ca2+ buffer systems that limit the diffusion of Ca2+ ions [5]. Ca2+ buffers—a class of cytosolic Ca2+-binding proteins—act as modulators of short-lived intracellular Ca2+ signals. Each neuron is equipped with proteins such as Ca2+ channels, transporters, pumps, and Ca2+ buffers that together shape the intracellular Ca2+ signals. A recent study demonstrated that the absence of Ca2+-binding proteins, such as parvalbumins, calbindins, and calretinins, leads to an increase in neuronal excitability through a reduction in cytosolic Ca2+-buffering capacity, suggesting a link between the expression of cellular Ca2+-binding proteins and excitability [6–8]. Alterations in the Ca2+-calmodulin kinase systems have been associated with some strains of seizure-susceptible mice, suggesting that calmodulin-mediated processes may play a role in the development of altered neuronal excitability and in some forms of seizure disorders [9].
Bouts of action potentials can lead to an increase in the cytosolic Ca2+ concentration of neurons through the activation of voltage-gated Ca2+ channels (VGCCs) or Ca2+-permeable transient receptor potential (TRP) channels, as well as through the release of Ca2+ from intracellular stores via Ca2+-induced Ca2+ release mechanisms. As a result, increased cytosolic Ca2+ levels would control neuronal excitability by modulating many ion channels, including Ca2+-activated K+ (KCa) channels or Ca2+-activated TRP channels, which regulate the firing activities and patterns of SNc dopamine neurons [10, 11]. In midbrain dopaminergic neurons, the steep Ca2+ concentration gradient enables it to cross the plasma membrane into neurons through open pores such as L-type VGCCs, which appear to play a major role in regulating firing activities and intracellular Ca2+ concentration dynamics [12, 13]. Ca2+ is also able to flow into the cytoplasm though the inositol trisphosphate receptor (IP3R) and ryanodine receptor (RyR) ion channels in the ER membrane [14]. The influx of Ca2+ via VGCCs or IP3R/RyR in a depolarized state leads to the activation of small-conductance KCa (SK) channels. This generates a large afterhyperpolarization that affects the interspike interval of pacemaker firing in the dopaminergic midbrain neurons [15]. It was recently reported that many types of TRP channels are widespread in midbrain dopamine neurons [16, 17] and that they play a critical role in Ca2+ homeostasis and the regulation of firing in dopaminergic SNc neurons [10].
In light of these recent reports, this study was undertaken to examine how Zn2+ contributes to altered neuronal excitability in dopaminergic SNc neurons, depending on the cytosolic Ca2+ capacity and Ca2+-related channel processes, using a whole-cell patch recording in SNc neurons. We found that SK channel activity, which is influenced by changes in the cytosolic Ca2+ concentration under regulation by the Ca2+-buffering capacity, was essential in the Zn2+-mediated alteration of firing activity.
MATERIALS AND METHODS
Brain preparation
All experiments followed the guidelines issued by Ewha Womans University on the ethical use of animals for experimentation.
The rats (Sprague-Dawley, age 11~20 days) were anesthetized with isoflurane and decapitation. The brain was quickly removed from the rat skull and immersed in ice-cold high-concentration sucrose solution containing (in mM): 201 sucrose, 3 KCl, 1.25 NaH2PO4, 2 MgCl2, 2 CaCl2, 26 NaHCO3, and 10 D-glucose. Coronal midbrain slice (300 μm) containing SNC was cut in ice-cold high-concentration sucrose solution using a vibratome (VT1000S, Leica Microsystems, Germany). Before recording, slices were incubated at 30°C for 60 min in artificial cerebrospinal fluid (ACSF: 95% O2/5% CO2) containing (in mM): 126 NaCl, 3 KCl, 1.25 NaH2PO4, 1.3 MgSO4, 2.4 CaCl2, 26 NaHCO3, and 10 D-glucose. After the recovery period, slices were maintained in ACSF at room temperature (20±3°C).
Dissociated SNc dopamine neurons were prepared from rats (Sprague-Dawley, age 9~13 days) as described previously [4]. Whole brains from decapitated rats were quickly removed and placed in an ice-cold oxygenated N-2-hydroxyethylpiperazine-N’-2-ethanesulphonic acid (HEPES)-buffered saline containing (in mM): 135 NaCl, 5 KCl, 1 CaCl2, 1 MgCl2, 25 D-glucose, 10 HEPES (pH 7.3). Coronal slice (300~400 μm) containing SNc was cut using a vibratome (VT1000S, Leica). The dissected SNc segments were incubated in a fully oxygenated HEPES-buffered saline containing papain (10 U/ml; Worthington Biochemical Corp., Lakewood, NJ, USA) for 30 min at 36°C. Tissue segments were rinsed with enzyme-free saline and mildly triturated with a graded series of fire-polished Pasteur pipettes. Dissociated SNC neurons were plated onto 35-mm Petri-dishes at room temperature (20±3°C).
Electrophysiological recordings
Whole-cell current clamp records were obtained from dopaminergic neurons in SNc slices as previously described [3]. Recordings were performed in a submersion-type chamber (2~4 ml/min perfusion of oxygenated modified ACSF at 30~32°C) mounted on an upright microscope (Olympus BX51WI, Tokyo, Japan). Patch pipettes (3~4 MΩ) were pulled on a Narishige electrode puller PP-83 (Narishige, Tokyo, Japan) from Kimax borosilicate glass capillaries (KG-33, Precision Glass, Inc., Claremont, CA, USA) with an inner diameter of 1.2 mm and an outer diameter of 1.5 mm. The internal electrode solution contained (in mM): 115 K-gluconate, 10 HEPES, 1 MgCl2, 10 KCl, 1 CaCl2, 11 EGTA or 11 BAPTA, 4 Na2-ATP (pH 7.3). Spikes in current-clamp mode were acquired with a multiclamp700B amplifier (Molecular Devices, Sunnyvale, CA, USA) and a Digidata-1440A A/D converter (Molecular Devices) using pClamp10 (Molecular Devices). Currents were Bessel-filtered at a cut-off frequency of 5 kHz and digitally sampled at 10 kHz. Data graphing and response analyses were performed with the GraphPad Prism8.0 (GraphPad Software Inc., La Jolla, CA, USA) and pClamp10 (Molecular Devices).
Whole-cell recordings were obtained from isolated SNc using an Axopatch 200B amplifier (Molecular Devices, Sunnyvale, CA, USA) with a Digidata-1320A/D converter and analyzed using pClamp9 software (Molecular Devices). The Patch pipettes resistances were 3.0~3.5 MΩ. Recording pipettes were filled with (in mM): 135 CsCl, 2 MgCl2, 10 HEPES, 11 EGTA, 4 ATP-Na2 (pH 7.3). Series resistances were compensated (>50%) and monitored at regular intervals. Leakage and capacitive currents were subtracted on-line from active responses using P/5 protocols. Data graphing and response analyses were performed with the Graph-Pad Prism8.0 (GraphPad Software Inc.) and pClamp9 (Molecular Devices).
Pipettes were pulled from standard wall borosilicate glass capillaries (o.d., 1.5 mm; i.d.,1.2 mm; KG-33, Garner Glass) using a two-stage puller (PP-83, Narishige) and coated with sylgard (Dow Corning) or Sigmacote (Sigma) to reduce electrical noise. Pipette resistance was 10~20 MΩ. The data were filtered at 2 kHz (four-pole Bassel) and digitized at 20 kHz. Channel open frequency was analyzed by the pClamp10 using a 50% threshold detection criterion. Events briefer than 150 μs were ignored.
Solutions
For the current-clamp recordings of slices, a modified ACSF, in which NaH2PO4 was omitted and MgSO4 was replaced with MgCl2, was used as an extracellular solution to avoid any Zn2+-related precipitates. For the measurement of voltage-gated Ca2+ channels in a voltage-clamp mode, the composition of bath solution in control was (in mM): 155 tetraethylammonium chloride, 10 BaCl2, and 10 HEPES (pH 7.4). Bath solutions were added to the recording chamber, using a perfusion system that consisted of 5 polyethylene tubes arranged in parallel in one plane for rapid exchange. All chemicals were purchased from Sigma-Aldrich (St Louis, MO, USA) and Tocris (Ellisville, MO, USA). For inside-out patch single-channel recordings of SK channels, bath and pipettes contained (mM): 140 KCl, 10 HEPES, 5 MgCl2, pH 7.2. To activate SK channels, free calcium concentration in the bath solution was adjusted by addition of 100 μM CaCl2.
Statistical analysis
Data are presented as mean±standard error mean (S.E.M). Statistical significance was estimated with two-tailed Student’s
RESULTS
Differential modulation of the excitability in SNc dopaminergic neurons by zinc, depending on internal calcium chelators
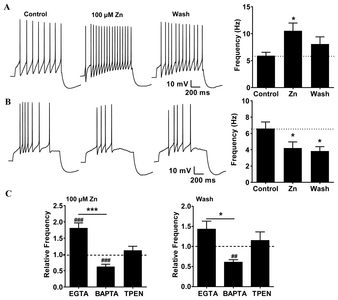
Dopaminergic SNc neurons recorded in juvenile rat brain slices were identified in a whole-cell patch-clamp by the presence of a prominent sag during hyperpolarizing current pulses, and repetitive firing due to anode break excitation, which are well-known criteria for identifying dopaminergic neurons in the SNc [4, 18, 19]. We had previously reported seemingly contradictory Zn2+-mediated effects on the excitability of SNc neurons, where Zn2+ increased the excitability of dopaminergic neurons acutely isolated from the rat SNc [4], but decreased the firing activity in the dopaminergic neurons of SNc slices [3].
To investigate the influence of the Ca2+-buffering capacity on Zn2+-mediated changes in the excitability of SNc neurons, we examined the effects of Zn2+ on the activity patterns of SNc neurons that had received depolarizing pulses through EGTA- or BAPTA-containing intracellular solutions in a pipette. When the pipette contained EGTA, the evoked action potentials revealed regularity in the spike frequency patterns ( A, Control). Zn2+ (100 μM) significantly increased the spike frequency to 181% (Fig. 1C, 100 μM Zn2+), but this effect was not completely reversed after the washout (Fig. 1C, Wash). However, when using the same concentration of BAPTA, the pattern of firing activities evoked by depolarization showed the opposite effect, suggesting that the Ca2+-buffering capacity might alter the Zn2+-induced modulation of excitability. The appearance of spike-frequency adaptation patterns in the pipettes containing BAPTA (Fig. 1B) suggested that the spike-frequency adaptation in SNc dopaminergic neurons occurs due to the lowering of cytoplasmic Ca2+ levels, which may cause a decrease in the activity of SK channels [20, 21]. In experiments with BAPTA-containing pipettes, Zn2+ significantly decreased the spike frequency to 63% (Fig. 1C, 100 μM Zn2+), but the firing activity did not recover completely after washout (Fig. 1C, Wash). BAPTA is also able to chelate Zn2+ influxes into the cytoplasm (KD values: Ca2+, 160×10−9 M; Zn2+, 8×10−9 M)[22]. Therefore, we examined the effects of internal Zn2+ on spike frequencies using pipettes containing the Zn2+ chelator N,N,N’,N’-tetrakis(2-pyridylmethyl) ethylenediamine (TPEN, 50 μM), which has a very high affinity for Zn2+ (KD=2.6×10−16 M), but relatively low affinities for Ca2+ (KD=4.0×10−5 M) and Mg2+ (KD=2.0×10−2 M) [23]. In experiments with TPEN-loaded pipettes, the Zn2+-mediated increase in excitability was less pronounced than in the EGTA experiments (Fig. 1C), suggesting that the blocking of Zn2+-mediated potentiation of spike activity may be due to the chelation of intracellular Zn2+ by both BAPTA and TPEN. However, we were unable to rule out the possibility that the membrane-permeable TPEN also chelates extracellular Zn2+, and we could not explain why the presence of TPEN does not result in reduced activity via enhanced IA. Therefore, we also tested the effect of extracellular Zn2+-mediated augmentation of excitability on Ca2+ channels in the cell membrane and the endoplasmic reticulum (ER).
Involvement of VGCCs and intracellular calcium stores in the zinc-mediated augmentation of excitability
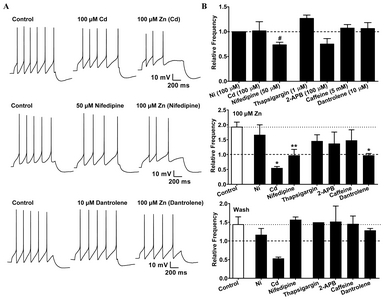
In SNc neurons, bouts of action potential can increase the cytosolic Ca2+ concentration either through the activation of VGCCs, or through the release of Ca2+ from intracellular stores via Ca2+-induced Ca2+ release signaling. The abrupt change in conductance enables Ca2+ to enter the neuronal cytoplasm rapidly through the open pores of L-type VGCCs in SNc dopaminergic neurons [24, 25]. First, we examined the changes in excitability in the presence of Cd2+ and Ni2+ to determine whether high-threshold VGCCs and low-threshold VGCCs are involved in the observed Zn2+-induced increases in excitability. In the presence of Cd2+ (100 μM), the Zn2+-induced increases in firing activities were completely blocked, and similar to the effect of BAPTA, this phenomenon did not recover after Zn2+ washout (Fig. 2A,
We further examined whether the release of Ca2+ from intracellular stores is responsible for the Zn2+-induced potentiation of excitability. To deplete intracellular Ca2+ stores, the Ca2+-ATPase inhibitor thapsigargin (1 μM) was added to the internal recording solution. Thapsigargin had no significant effects on firing activities, either in the presence of Zn2+, or after the Zn2+ washout (Fig. 2B). This suggested that Ca2+ release from the intracellular stores is not responsible for the Zn2+-induced potentiation of excitability. However, because thapsigargin leads to an increase in intracellular free Ca2+ by potently inhibiting the ER Ca2+-ATPase, we also tested the channels responsible for this Ca2+-induced Ca2+ release from the ER. Two major pathways have been identified for the release of Ca2+ from intracellular stores: the IP3- and ryanodine-sensitive Ca2+ stores [26]. To assess the potential role of IP3Rs in the Zn2+-mediated increase in firing frequency, we used a membrane-permeable IP3R antagonist, 2-aminoethoxydiphenyl borate (2-APB, 100 μM), which caused a 25% reduction in spike frequency but did not alter the Zn2+-mediated enhancement of firing activities. The release of Ca2+ from IP3Rs or the influx of Ca2+ via VGCCs could further trigger Ca2+-induced Ca2+ release from the RyRs. To examine the possible role of RyRs in Zn2+-mediated increases in activity, we used caffeine to activate the RyR-sensitive Ca2+ stores [27]. For these experiments, we used 5 mM caffeine, which exceeds the KD of 1.5 mM for caffeine-induced Ca2+ release from ER stores [28]. Caffeine had no significant effects on firing activities, either in the presence of Zn2+, or after Zn2+ washout (Fig. 2B). Dantrolene, which is a selective membrane-permeable inhibitor of RyRs [29, 30], inhibited the Zn2+-induced increase in spike frequency, but had no effect on firing activity after the Zn2+ washout (Fig. 2A, bottom; Fig. 2B). These findings indicated that the release of Ca2+ from the RyRs is involved in the augmentation of firing frequency in the presence of Zn2+.
Involvement of SK channels in the zinc-mediated augmentation of excitability
BAPTA and EGTA differ considerably in their Ca2+ binding rate constants, with BAPTA being about 150 times faster than EGTA [31]. Therefore, BAPTA is noticeably more effective in preventing the diffusion of free Ca2+ away from the entrance site at the plasma membrane. To determine whether the KCa channels (large-conductance KCa channels, BK channel; small-conductance KCa channels, SK channel), which are controlled by local Ca2+ signaling domains, are involved in the Zn2+-mediated modulation of excitability in SNc neurons, we examined the effects of BK and SK channels on neuronal excitability in the presence of Zn2+.
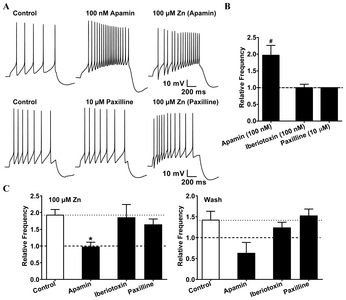
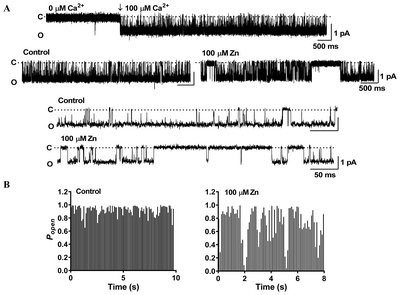
The SNc region has low expression of BK channels [32], and shows higher expression levels of the SK3 subunit [33, 34]. In our experiment, apamin (100 nM),—a highly specific blocker of SK channels—increased the excitability in dopaminergic neurons, whereas BK channel blockers like iberiotoxin (100 nM) and paxilline (10 μM) did not induce any changes in excitability (Fig. 3B). These observations supported previous reports [32–34] that apamin-sensitive SK channels are mainly expressed in SNc neurons, while BK channels are not. To elucidate the effect of Zn2+ on SK channels, we investigated whether the Zn2+-mediated increase in firing activity could be blocked by a pretreatment of apamin. Apamin pretreatment significantly attenuated the Zn2+-induced augmentation of excitability, while the BK channel blockers iberiotoxin and paxilline had no effect on it (Fig. 3C). The effects of apamin pretreatment were maintained even after the washout of extracellular Zn2+, suggesting that Zn2+ could be acting on an intracellular domain of the SK channels. These results indicated that like apamin, Zn2+ was capable of blocking the SK channels, thus inducing an increase in excitability. To confirm the possibility of inhibition of SK channels by Zn2+ in SNc neurons, we performed inside-out patch single-channel recording. We found that Zn2+ reduced the channel open probability of SK channel, suggesting that Zn2+ directly blocks inside SK channels (Fig. 4).
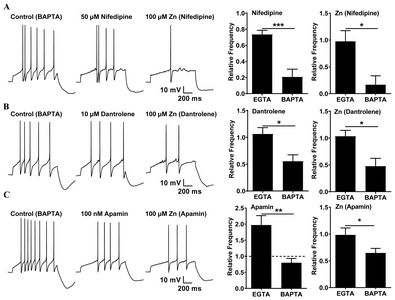
In order to determine how the effect of Zn2+ is different in the presence of BAPTA versus EGTA, the aforementioned experiments were conducted with BAPTA as well. Interestingly, in the presence of nifedipine or dantrolene, neuronal activities were lower in the BAPTA condition than in the EGTA condition (Fig. 5A and 5B), and increased neuronal activities were observed only when the SK channels were blocked by apamin in the EGTA condition (Fig. 5C). Moreover, nifedipine, dantrolene, and apamin significantly decreased the neuronal activity of Zn2+ under the BAPTA condition. Apamin increased neuronal activity under the EGTA condition and decreased it under the BAPTA condition, in a manner identical to the actions of Zn2+ (Fig. 5C).
Involvement of TRPM channels in the zinc-mediated augmentation of excitability
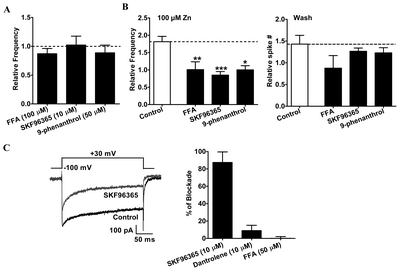
The tonic Ca2+ entry pathways that maintain basal cytosolic Ca2+ levels consist mainly of TRP channels, and contribute to the generation of spontaneous firings [10]. Therefore, the dopaminergic neurons in which Zn2+ enhanced firing activities were first tested for their response to flufenamic acid (FFA), a broad-spectrum TRP channel blocker. The application of FFA (100 μM) had no effect on firing activity, but it completely prevented the Zn2+-induced increase in firing activity (Fig. 6), suggesting that the potentiation of firing activity by Zn2+ occurred via TRP channels. On application of 10 μM of SKF96365—a widely-used blocker of canonical TRP (TRPC) channels—the potentiation of firing activity by Zn2+ was strongly inhibited (Fig. 6), but these effects were reversed upon the washout of SKF96365. In some preparations, SKF96365 also blocks VGCCs [35]. To determine whether the antagonists for Ca2+-related channels directly affect VGCCs, we tested SKF96365, dantrolene, and FFA on the VGCCs of acutely isolated SNc neurons using voltage-clamp recording (Fig. 6C). VGCC currents were elicited from isolated SNc neurons held at −100 mV by applying +30 mV, which activated all the VGCCs as expected. SKF96365 (10 μM) substantially reduced the steady-state VGCC currents by 87%, and dantrolene (10 μM) reduced the steady-state VGCC currents by 9%, but FFA (50 μM) did not significantly inhibit whole-cell steady-state VGCC currents (Fig. 6C). Therefore, we concluded that the inhibition of the Zn2+-induced firing potentiation by SKF96365 is mainly mediated via a VGCC inhibition, not a TRPC inhibition.
Melastatin-like TRP (TRPM) channels, which are directly activated by intracellular Ca2+, and conduct Na+ ions to further depolarize cells [36], are expressed in the neurons of the SNc [37]. To examine the possibility that TRPM channels are involved in the Zn2+-induced potentiation of excitability in SNc neurons, we applied 9-phenanthrol, a drug that selectively blocks TRPM channels [38]. 9-phenanthrol (50 μM) completely eliminated the Zn2+-induced amplification of firing activity in the tested neurons. These effects were reversed upon the washout of 9-phenanthrol, suggesting that TRPM channels could play an important role in the enhancement of firing activity by Zn2+.
DISCUSSION
Zn2+ is present in high concentration in the SN region of the parkinsonian brain [39–41], and can increase or inhibit the intrinsic firing activity of rat SNc dopaminergic neurons depending on the intracellular Ca2+ buffering capacity, suggesting the involvement of cytosolic Ca2+-dependent signaling processes. To elucidate the effect of Zn2+ on the excitability of dopaminergic SNc neurons, this study investigated its action on cytosolic Ca2+-dependent processes in relation to the intracellular Ca2+ buffering capacity.
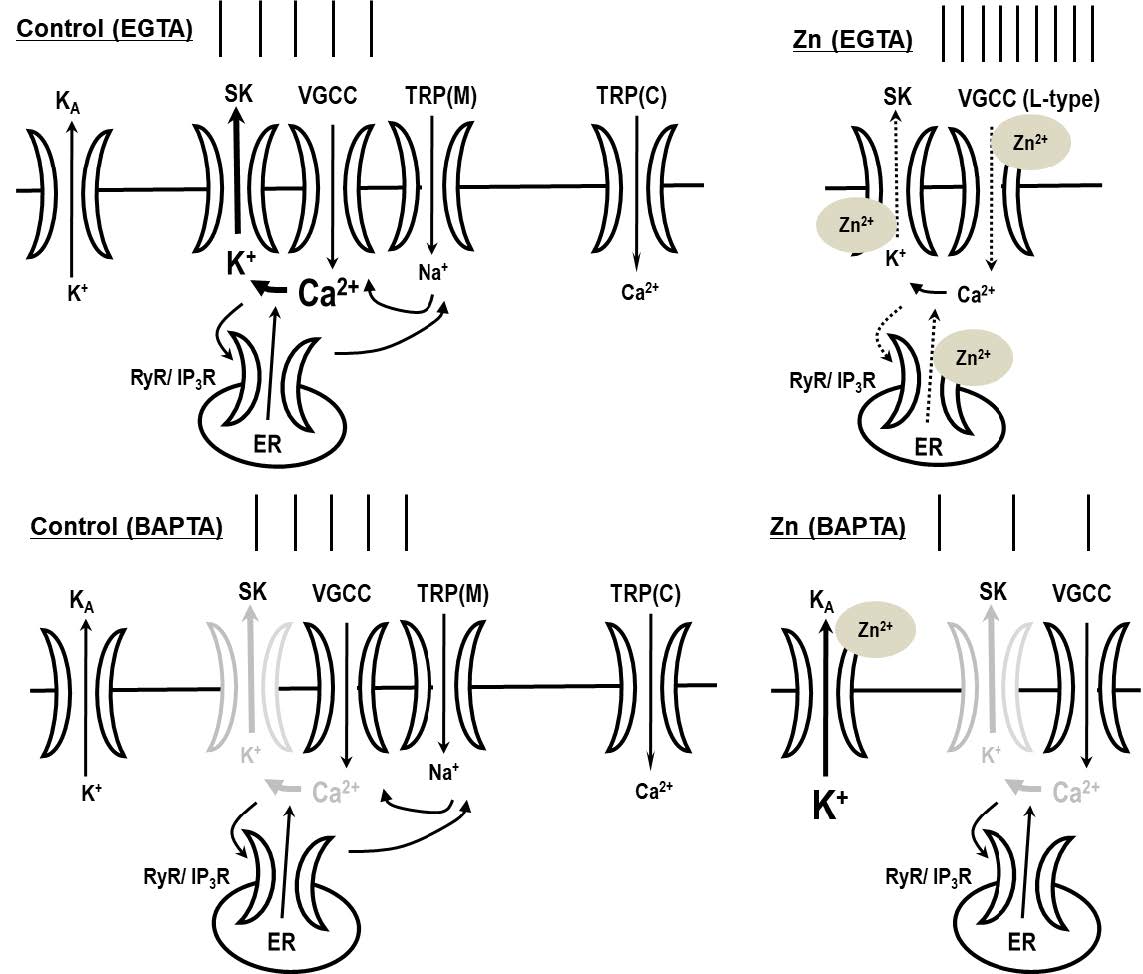
The prominent results of this study can be summarized as follows: (1) In the depolarized state, Zn2+ increases the tonic activity during intracellular dialysis with EGTA, but decreases firing activity in the presence of BAPTA. (2) The augmentation of firing activity in the presence of Zn2+ is blocked by the RyR blocker dantrolene, and by the VGCC antagonists nifedipine and Cd2+, but not by the IP3R antagonist 2-APB. (3) The Zn2+-mediated potentiation of firing activity is eliminated by the blockade of TRPM channels with FFA and 9-phenanthrol. (4) Zn2+ directly reduced the channel open probability of SK channels. Apamin—an SK channel inhibitor—enhances the firing activity, and its effect is attenuated by intracellular dialysis with BAPTA. Apamin blocks the augmentation of firing activity not only in the presence of Zn2+, but also after the washout of Zn2+.
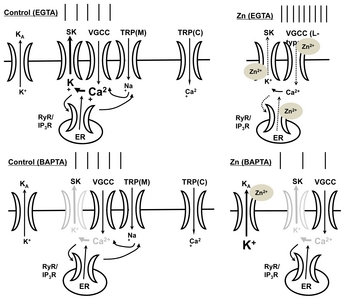
We propose some hypothetical mechanisms for Zn2+-mediated augmentation of excitability leading to Ca2+ signaling processes (Fig. 7). When SK channels are blocked, the firing activity of neurons can increase. Apamin is very similar in its actions compared to Zn2+. Apamin enhances tonic activity in the depolarized state when the cell is intracellularly dialyzed with EGTA, whereas apamin attenuates the firing activity in the presence of intracellular BAPTA (Fig. 5C). This is in agreement with a high expression of SK3 channels, but not BK channels in SNc neurons [32, 34]. Iberiotoxin and paxilline, which are BK channel antagonists, have no effect on the firing activity in SNc neurons (Fig. 3C). In the presence of apamin, the Zn2+-induced enhancement of firing activity is completely blocked and this blockage is sustained even after Zn2+ removal from the bath solution, suggesting that Zn2+ possibly acts on SK channels in both the extracellular and intracellular domains. In inside-out patch single-channel recording, Zn2+ directly reduced the SK channel open probability (Fig. 4). These results are consistent with the previous studies that Zn2+ has been reported to directly inhibit the activation of KCa channels in other preparations such as hippocampal neurons and motor neurons [42, 43]. Therefore, we suggest that Zn2+ enhances the firing activity of SNc neurons via the blockade of SK channels by Zn2+.
Cytosolic Ca2+ triggers a wide variety of Ca2+-dependent signaling events and reaction cascades, and its levels rise only for short periods of time and at spatially restricted domains. Such local Ca2+ signaling domains are generated by a variety of Ca2+ buffer systems, such as BAPTA and EGTA, which limit the diffusion of Ca2+ ions that have entered the cells through VGCCs or Ca2+-permeable channels [44, 45]. BAPTA, but not EGTA, interferes effectively with Ca2+ signaling processes in Ca2+ nanodomains, i.e., within 20~50 nm of the Ca2+ source, while both modulate Ca2+ signaling processes equally in Ca2+ microdomains, i.e. between 50 nm to a few hundred nm from the Ca2+ source [5]. The ability of Zn2+ to enhance the excitability in the presence of EGTA, but not of BAPTA, suggests that rapid local changes in Ca2+ are necessary for the augmentation of firing activity. In SNc neurons, one potential mechanism for rapid Ca2+ dynamics causing Zn2+-mediated increases in firing activity is the co-localization of VGCCs or RyRs as Ca2+ sources, and SK channels as Ca2+ sensors. If those VGCCs or RyRs and SK channels were located in Ca2+ nanodomains, Zn2+ would have no effect on neuronal excitability when using a BAPTA-loaded pipette, because BAPTA would rapidly decrease the free Ca2+ concentration, thus inactivating the SK channel. However, in the presence of EGTA, the reduction of SK channel activation by Zn2+ results in an augmentation of excitability. Because Zn2+ increases the excitability in experiments with an EGTA-containing, but not a BAPTA-containing, internal solution, we suggest that VGCCs or RyRs and SK channels are located in Ca2+ nanodomains, and Zn2+ possibly attenuates SK channel activity. In the BAPTA condition, due to a decrease in the levels of free Ca2+, Zn2+-induced neuronal activity would be determined by the action of KA channels (Fig. 7).
Because the Zn2+-mediated potentiation of firing activity in SNc neurons was completely blocked by pretreatment with nifedipine, Cd2+, or dantrolene (Fig. 2), we conclude that the Zn2+-mediated augmentation of activity requires an increase in cytosolic Ca2+ concentration via the activation of L-type VGCCs and RyRs. Zn2+ may directly block the SK channels, and/or indirectly reduce SK channel activity via a blockade of Ca2+ channels such as the VGCC or RyR/IP3R, thus attenuating the cytosolic Ca2+ concentration. Zn2+ significantly reduces the VGCC currents of SN neurons [46]. A plausible explanation for the observed Zn2+-induced potentiation of neuronal excitability is the indirect inhibition of SK channels caused by a decrease in Ca2+ influx due to the inhibition of VGCCs or RyRs by Zn2+.
TRPM channels, which are activated by cytosolic Ca2+ released from IP3Rs on the ER membrane, further depolarize the neurons by conducting Na+ ions, and induce a Ca2+ influx by activating the VGCCs. When applied extracellularly, 2-APB—a membrane-permeable blocker of intracellular IP3Rs—augments Ca2+ entry through store-operated channels (SOCs) at lower concentrations (<10 μM), and blocks TRPC channels at higher concentrations (>30 μM) [47–51]. The presence of 100 μM 2-APB failed to inhibit the Zn2+-mediated augmentation of firing activity, suggesting that the potentiation of neuronal activity by Zn2+ is not mediated by IP3Rs and TRPC channels. SKF96365, another well-known antagonist of TRPC channels, blocks SOCs and TRPC channels at concentrations of 25~100 μM [52–54], and in some preparations also blocks VGCCs [35]. SKF96365 was a potent blocker of VGCCs in this study as well (Fig. 4C). Because VGCC antagonists, such as nifedipine or Cd2+ (Fig. 2B), completely blocked the Zn2+-mediated increase in firing activity, we suggest that the prevention of Zn2+ action by SKF96365 is exclusively mediated by VGCC inhibition, and not by the blockade of TRPC channels. 9-phenanthrol blocks TRPM4 channels in a human embryonic kidney cell expression system with a half-maximal inhibitory concentration (IC50) of 16.7 μM under conventional whole-cell patch-clamp conditions, and does not alter the activity of TRPC channels, BK channels, inward-rectifying K+ channels, or VGCCs [55]. Therefore, FFA and 9-phenanthrol halt the Zn2+-mediated potentiation of firing activity by inhibiting the TRPM—most likely, TRPM4—channels. We suggest that the Zn2+-mediated inhibition of TRPM channels causes a decrease in SK activity via the reduction of cytosolic Ca2+, which contributes to the observed augmentation of firing activity in the SNc neurons.
In summary, we propose that (1) extracellular Zn2+ reduces the cytosolic Ca2+ concentration not only via the blockade of VGCCs, but also by reducing the Ca2+-mediated inactivation of RyRs/TRPM channels, resulting in a decrease in the activation of SK channels, and (2) Zn2+ directly blocks SK channels via interactions with an intracellular domain. This suggests that in the SNc, the blockade of SK channels by Zn2+ directly and/or indirectly induces the Zn2+-mediated augmentation of excitability.
Several Ca2+-binding proteins—or Ca2+ buffering proteins, including calretinin, calbindin and parvalbumin—are found throughout the central nervous system, including in the midbrain dopaminergic neurons, and help maintain a tight regulation of Ca2+ signals [56–58]. These high-affinity Ca2+-binding proteins act as buffers to prevent abrupt changes in the intracellular Ca2+ concentration [59, 60], and this is essential in preventing cell dysfunction and excitotoxic damage caused by high intracytoplasmic concentrations of Ca2+ [49, 61]. Some reports emphasize the role of Ca2+-sensing receptor or Ca2+-binding proteins/ Ca2+-buffering protein in pathophysiology of glaucoma and vulnerability to traumatic noise [62, 63]. The pathology of Parkinson’s disease involves a significant loss of parvalbumin-immunoreactivity in SN neurons [64], suggesting that this Ca2+-buffering protein plays a crucial role in maintaining normal Ca2+ signaling processes and Ca2+ homeostasis. Based on the results our experiments using EGTA and BAPTA, we propose that Zn2+ could increase the excitability of SNc neurons in pathological conditions involving a decrease in the activity of Ca2+-binding proteins, similar to the EGTA condition, whereas Zn2+ could decrease the firing activity in physiological conditions involving an increased activity of Ca2+-binding proteins, similar to BAPTA condition. This suggests that exogenous or endogenous factors such as Zn2+ could differentially modulate the excitability of neurons, depending on the physiological or pathological state of synaptic as determined by the Ca2+-buffering capacity. By attenuating Ca2+ influx into the cytosol, Zn2+ may also compensate for the loss of SNc dopaminergic neurons during the development of Parkinson’s disease [65], thus maintaining normal information processes in the basal ganglia until a critical mass of dopaminergic neurons are generated.
ACKNOWLEDGEMENTS
The present research was supported by the research fund of Dankook university in 2017.
Figures
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
References
- Eide DJ (2006) Zinc transporters and the cellular trafficking of zinc. Biochim Biophys Acta 1763:711-722.


- Takeda A (2000) Movement of zinc and its functional significance in the brain. Brain Res Brain Res Rev 34:137-148.
- Noh J, Chang SY, Wang SY, Chung JM (2011) Dual function of Zn2+ on the intrinsic excitability of dopaminergic neurons in rat substantia nigra. Neuroscience 175:85-92.
- Chung J, Chang S, Kim Y, Shin H (2000) Zinc increases the excitability of dopaminergic neurons in rat substantia nigra. Neurosci Lett 286:183-186.
- Fakler B, Adelman JP (2008) Control of K(Ca) channels by calcium nano/microdomains. Neuron 59:873-881.
- Foehring RC, Zhang XF, Lee JC, Callaway JC (2009) Endogenous calcium buffering capacity of substantia nigral dopamine neurons. J Neurophysiol 102:2326-2333.

- Roussel C, Erneux T, Schiffmann SN, Gall D (2006) Modulation of neuronal excitability by intracellular calcium buffering: from spiking to bursting. Cell Calcium 39:455-466.
- Schwaller B (2010) Cytosolic Ca2+ buffers. Cold Spring Harb Perspect Biol 2:a004051.
- DeLorenzo RJ (1984) Calmodulin systems in neuronal excitability: a molecular approach to epilepsy. Ann Neurol 16 Suppl:S104-S114.
- Kim SH, Choi YM, Jang JY, Chung S, Kang YK, Park MK (2007) Nonselective cation channels are essential for maintaining intracellular Ca2+ levels and spontaneous firing activity in the midbrain dopamine neurons. Pflugers Arch 455:309-321.
- Shepard PD, Bunney BS (1991) Repetitive firing properties of putative dopamine-containing neurons in vitro: regulation by an apamin-sensitive Ca(2+)-activated K+ conductance. Exp Brain Res 86:141-150.
- Cardozo DL, Bean BP (1995) Voltage-dependent calcium channels in rat midbrain dopamine neurons: modulation by dopamine and GABAB receptors. J Neurophysiol 74:1137- 1148.
- Durante P, Cardenas CG, Whittaker JA, Kitai ST, Scroggs RS (2004) Low-threshold L-type calcium channels in rat dopamine neurons. J Neurophysiol 91:1450-1454.
- Chan CS, Gertler TS, Surmeier DJ (2009) Calcium homeostasis, selective vulnerability and Parkinson's disease. Trends Neurosci 32:249-256.
- Wolfart J, Roeper J (2002) Selective coupling of T-type calcium channels to SK potassium channels prevents intrinsic bursting in dopaminergic midbrain neurons. J Neurosci 22:3404-3413.
- De March Z, Giampà C, Patassini S, Bernardi G, Fusco FR (2006) Cellular localization of TRPC5 in the substantia nigra of rat. Neurosci Lett 402:35-39.
- Guatteo E, Chung KK, Bowala TK, Bernardi G, Mercuri NB, Lipski J (2005) Temperature sensitivity of dopaminergic neurons of the substantia nigra pars compacta: involvement of transient receptor potential channels. J Neurophysiol 94:3069-3080.
- Hajós M, Greenfield SA (1994) Synaptic connections between pars compacta and pars reticulata neurones: electrophysiological evidence for functional modules within the substantia nigra. Brain Res 660:216-224.
- Richards CD, Shiroyama T, Kitai ST (1997) Electrophysiological and immunocytochemical characterization of GABA and dopamine neurons in the substantia nigra of the rat. Neuroscience 80:545-557.
- Ha GE, Cheong E (2017) Spike frequency adaptation in neurons of the central nervous system. Exp Neurobiol 26:179-185.
- Vandecasteele M, Deniau JM, Venance L (2011) Spike frequency adaptation is developmentally regulated in substantia nigra pars compacta dopaminergic neurons. Neuroscience 192:1-10.
- Grynkiewicz G, Poenie M, Tsien RY (1985) A new generation of Ca2+ indicators with greatly improved fluorescence properties. J Biol Chem 260:3440-3450.
- Stork CJ, Li YV (2006) Intracellular zinc elevation measured with a "calcium-specific" indicator during ischemia and reperfusion in rat hippocampus: a question on calcium overload. J Neurosci 26:10430-10437.
- Chan CS, Guzman JN, Ilijic E, Mercer JN, Rick C, Tkatch T, Meredith GE, Surmeier DJ (2007) 'Rejuvenation' protects neurons in mouse models of Parkinson's disease. Nature 447:1081-1086.
- Nedergaard S, Flatman JA, Engberg I (1993) Nifedipine- and omega-conotoxin-sensitive Ca2+ conductances in guinea-pig substantia nigra pars compacta neurones. J Physiol 466:727-747.
- Berridge MJ (1998) Neuronal calcium signaling. Neuron 21:13-26.
- Rousseau E, Meissner G (1989) Single cardiac sarcoplasmic reticulum Ca2+-release channel: activation by caffeine. Am J Physiol 256:H328-H333.
- Hoesch RE, Weinreich D, Kao JP (2001) A novel Ca(2+) influx pathway in mammalian primary sensory neurons is activated by caffeine. J Neurophysiol 86:190-6.
- Fruen BR, Mickelson JR, Louis CF (1997) Dantrolene inhibition of sarcoplasmic reticulum Ca2+ release by direct and specific action at skeletal muscle ryanodine receptors. J Biol Chem 272:26965-26971.
- Zhao F, Li P, Chen SR, Louis CF, Fruen BR (2001) Dantrolene inhibition of ryanodine receptor Ca2+ release channels. Molecular mechanism and isoform selectivity. J Biol Chem 276:13810-13816.
- Naraghi M, Neher E (1997) Linearized buffered Ca2+ diffusion in microdomains and its implications for calculation of [Ca2+] at the mouth of a calcium channel. J Neurosci 17:6961-6973.
- Sausbier U, Sausbier M, Sailer CA, Arntz C, Knaus HG, Neuhuber W, Ruth P (2006) Ca2+ -activated K+ channels of the BK-type in the mouse brain. Histochem Cell Biol 125:725-741.
- Sailer CA, Kaufmann WA, Marksteiner J, Knaus HG (2004) Comparative immunohistochemical distribution of three small-conductance Ca2+-activated potassium channel subunits, SK1, SK2, and SK3 in mouse brain. Mol Cell Neurosci 26:458-469.
- Stocker M, Pedarzani P (2000) Differential distribution of three Ca(2+)-activated K(+) channel subunits, SK1, SK2, and SK3, in the adult rat central nervous system. Mol Cell Neurosci 15:476-493.
- Leung YM, Kwan CY (1999) Current perspectives in the pharmacological studies of store-operated Ca2+ entry blockers. Jpn J Pharmacol 81:253-258.
- Ullrich ND, Voets T, Prenen J, Vennekens R, Talavera K, Droogmans G, Nilius B (2005) Comparison of functional properties of the Ca2+-activated cation channels TRPM4 and TRPM5 from mice. Cell Calcium 37:267-278.
- Mrejeru A, Wei A, Ramirez JM (2011) Calcium-activated non-selective cation currents are involved in generation of tonic and bursting activity in dopamine neurons of the substantia nigra pars compacta. J Physiol 589(Pt 10):2497-2514.
- Grand T, Demion M, Norez C, Mettey Y, Launay P, Becq F, Bois P, Guinamard R (2008) 9-phenanthrol inhibits human TRPM4 but not TRPM5 cationic channels. Br J Pharmacol 153:1697-1705.
- Dexter DT, Carayon A, Javoy-Agid F, Agid Y, Wells FR, Daniel SE, Lees AJ, Jenner P, Marsden CD (1991) Alterations in the levels of iron, ferritin and other trace metals in Parkinson's disease and other neurodegenerative diseases affecting the basal ganglia. Brain 114(Pt 4):1953-1975.
- Dexter DT, Wells FR, Lees AJ, Agid F, Agid Y, Jenner P, Marsden CD (1989) Increased nigral iron content and alterations in other metal ions occurring in brain in Parkinson's disease. J Neurochem 52:1830-1836.
- Tarohda T, Ishida Y, Kawai K, Yamamoto M, Amano R (2005) Regional distributions of manganese, iron, copper, and zinc in the brains of 6-hydroxydopamine-induced parkinsonian rats. Anal Bioanal Chem 383:224-234.
- Miralles F, Canti C, Marsal J, Peres J, Solsona C (1994) Zinc ions block rectifier potassium channels and calcium activated potassium channels at the frog motor nerve endings. Brain Res 641:279-284.
- Sim JA, Cherubini E (1990) Submicromolar concentrations of zinc irreversibly reduce a calcium-dependent potassium current in rat hippocampal neurons in vitro. Neuroscience 36:623-629.
- Augustine GJ, Santamaria F, Tanaka K (2003) Local calcium signaling in neurons. Neuron 40:331-346.
- Neher E (1998) Vesicle pools and Ca2+ microdomains: new tools for understanding their roles in neurotransmitter release. Neuron 20:389-399.
- Noh J, Chung JM (2000) Effect of zinc ions on voltage-gated Ca2+ currents of rat nigral neurons. Exp Neurobiol 9:25-28.
- Bakowski D, Glitsch MD, Parekh AB (2001) An examination of the secretion-like coupling model for the activation of the Ca2+ release-activated Ca2+ current I(CRAC) in RBL-1 cells. J Physiol 532(Pt 1):55-71.
- Gregory RB, Rychkov G, Barritt GJ (2001) Evidence that 2-aminoethyl diphenylborate is a novel inhibitor of storeoperated Ca2+ channels in liver cells, and acts through a mechanism which does not involve inositol trisphosphate receptors. Biochem J 354(Pt 2):285-290.
- Iwasaki H, Mori Y, Hara Y, Uchida K, Zhou H, Mikoshiba K (2001) 2-Aminoethoxydiphenyl borate (2-APB) inhibits capacitative calcium entry independently of the function of inositol 1,4,5-trisphosphate receptors. Receptors Channels 7:429-439.
- Ma HT, Venkatachalam K, Parys JB, Gill DL (2002) Modification of store-operated channel coupling and inositol trisphosphate receptor function by 2-aminoethoxydiphenyl borate in DT40 lymphocytes. J Biol Chem 277:6915-6922.
- Prakriya M, Lewis RS (2001) Potentiation and inhibition of Ca2+ release-activated Ca2+ channels by 2-aminoethyldiphenyl borate (2-APB) occurs independently of IP3 receptors. J Physiol 536(Pt 1):3-19.
- Boulay G, Zhu X, Peyton M, Jiang M, Hurst R, Stefani E, Birnbaumer L (1997) Cloning and expression of a novel mammalian homolog of Drosophila transient receptor potential (Trp) involved in calcium entry secondary to activation of receptors coupled by the Gq class of G protein. J Biol Chem 272:29672-29680.
- Halaszovich CR, Zitt C, Jungling E, Luckhoff A (2000) Inhibition of TRP3 channels by lanthanides. Block from the cytosolic side of the plasma membrane. J Biol Chem 275:37423- 37428.
- Zhu X, Jiang M, Birnbaumer L (1998) Receptor-activated Ca2+ influx via human Trp3 stably expressed in human embryonic kidney (HEK)293 cells. Evidence for a non-capacitative Ca2+ entry. J Biol Chem 273:133-142.
- Gonzales AL, Garcia ZI, Amberg GC, Earley S (2010) Pharmacological inhibition of TRPM4 hyperpolarizes vascular smooth muscle. Am J Physiol Cell Physiol 299:C1195-C1202.
- Celio MR (1990) Calbindin D-28k and parvalbumin in the rat nervous system. Neuroscience 35:375-475.
- Résibois A, Rogers JH (1992) Calretinin in rat brain: an immunohistochemical study. Neuroscience 46:101-134.
- Rogers JH (1992) Immunohistochemical markers in rat brain: colocalization of calretinin and calbindin-D28k with tyrosine hydroxylase. Brain Res 587:203-210.
- Chard PS, Bleakman D, Christakos S, Fullmer CS, Miller RJ (1993) Calcium buffering properties of calbindin D28k and parvalbumin in rat sensory neurones. J Physiol 472:341-357.
- Lledo PM, Somasundaram B, Morton AJ, Emson PC, Mason WT (1992) Stable transfection of calbindin-D28k into the GH3 cell line alters calcium currents and intracellular calcium homeostasis. Neuron 9:943-954.
- Kurobe N, Inaguma Y, Shinohara H, Semba R, Inagaki T, Kato K (1992) Developmental and age-dependent changes of 28- kDa calbindin-D in the central nervous tissue determined with a sensitive immunoassay method. J Neurochem 58:128-134.
- Sharma K, Seo YW, Yi E (2018) Differential expression of Ca2+-buffering protein calretinin in cochlear afferent fibers: a possible link to vulnerability to traumatic noise. Exp Neurobiol 27:397-407.
- Lee JY, Oh SJ (2018) Functional analysis and immunochemical analyses of Ca2+ homeostasis-related proteins expression of glaucoma-induced retinal degeneration in rats. Exp Neurobiol 27:16-27.
- Hardman CD, McRitchie DA, Halliday GM, Cartwright HR, Morris JG (1996) Substantia nigra pars reticulata neurons in Parkinson's disease. Neurodegeneration 5:49-55.
- Joe EH, Choi DJ, An J, Eun JH, Jou I, Park S (2018) Astrocytes, microglia, and Parkinson's disease. Exp Neurobiol 27:77-87.