Articles

Article Tools
 View Full Text View Full Text |
 Abstract Abstract |
 Article as PDF Article as PDF |
 Print this Article Print this Article |
 Pubmed Pubmed |
 PMC PMC |
 PubReader PubReader |
 Export to Citation Export to Citation |
 Email Alerts Email Alerts |
 Open Access Open Access |
Share this article on :



Stats or Metrics
Article
Original Article
Exp Neurobiol 2013; 22(4): 301-307
Published online December 30, 2013
https://doi.org/10.5607/en.2013.22.4.301
© The Korean Society for Brain and Neural Sciences
HMGB1-Binding Heptamer Confers Anti-Inflammatory Effects in Primary Microglia Culture
Il-Doo Kim and Ja-Kyeong Lee*
Department of Anatomy, Inha University School of Medicine, Incheon 400-712, Korea
Correspondence to: *To whom correspondence should be addressed.
TEL: 82-32-890-0913, FAX: 82-32-884-2105
e-mail: jklee@inha.ac.kr
High mobility group box 1 (HMGB1) is an endogenous danger signal molecule. In the postischemic brain, HMGB1 is massively released during NMDA-induced acute damage and triggers inflammatory processes. In a previous study, we demonstrated that intranasally delivered HMGB1 binding heptamer peptide (HBHP; HMSKPVQ) affords robust neuroprotective effects in the ischemic brain after middle cerebral artery occlusion (MCAO, 60 minutes). In the present study, we investigated HBHP-induced anti-inflammatory effects on microglia activation. In LPS-treated primary microglia culture, HMGB1 was rapidly released and accumulated in culture media. Furthermore, LPS-conditioned media collected from primary microglia cultures (LCM) activated naïve microglia and markedly induced NO and proinflammatory cytokines. However, the suppression of HMGB1 by siRNA-HMGB1, HMGB1 A box, or anti-HMGB1 antibody significantly attenuated LCM-induced microglial activation, suggesting that HMGB1 plays a critical role in this process. A pull-down assay using biotin-labeled HBHP showed that HBHP binds directly to HMGB1 (more specifically to HMGB1 A box) in LCM. In addition, HBHP consistently inhibited LCM-induced microglial activation and suppressed the inductions of iNOS and proinflammatory cytokines. Together these results suggest that HBHP confers anti-inflammatory effects in activated microglia cultures by forming a complex with HMGB1.
Keywords: HMGB1, HBHP, inflammation, microglia
INTRODUCTION
High mobility group box 1 (HMGB1) is an endogenous danger signal molecule, which is released by necrotic cells or actively secreted by macrophages and monocytes into the extracellular milieu, and induces inflammation [1, 2]. When released extracellularly, HMGB1 serves as a danger signal that evokes inflammatory reactions by activating various immune-related cells, including microglia in brain [3, 4]. Furthermore, extracellular HMGB1 upregulates inflammatory cytokines, such as, IL-1β, IL-6, and TNF-α, leading to the activations of macrophages/monocytes and the maturation of dendritic cells (DCs) [5, 6]. In accordance with these observations, high plasma levels of HMGB1 have been reported to be correlated with disease severity in various pathological conditions, including sepsis [7, 8], pancreatitis [9], and stroke [10].
HMGB1 release, which occurs under various pathological conditions in the brain, aggravates inflammatory processes [11]. In a previous report, we demonstrated that HMGB1 is massively released during N-methyl-D-aspartate (NMDA)-induced acute damage in the postischemic brain, and that it exacerbates neuronal damage and triggers inflammatory processes [3, 12]. Other studies have reported that the administration of HMGB1 monoclonal antibody suppresses infarct formation in the postischemic brain [13] and protects the blood-brain barrier [14]. Furthermore, siRNA-mediated HMGB1 knockdown and HMGB1 A box-mediated HMGB1 inhibition have both been reported to markedly reduce infarct volume in the brain of a rat middle cerebral artery occlusion (MCAO) model [15-17].
In a previous ligand screening study using a phage-displayed heptapeptide library, a number of heptamer peptides that binds to HMGB1 A box were identified [18]. In a previous study, we found that a HMGB1-binding heptamer peptide (HBHP) with the HMSKPVQ sequence, which directly binds with HMGB1 under cell free conditions and in NMDA-conditioned media (MCM), exerted a neuroprotective effect in the rat postischemic brain [19]. Moreover, intranasally delivered HBHP suppressed infarct formation in the rat brain after MCAO and ameliorated neurological deficits [19]. In the present study, we investigated whether HBHP confers anti-inflammatory effects on cultured primary microglia, and whether its effects are attributable to direct binding to HMGB1.
MATERIALS AND METHODS
Primary microglial cultures were prepared as previously described [3] (Kim et al., 2006). In brief, cells dissociated from the cerebral hemispheres of 1 day-old postnatal rat brains (Sprague-Dawley strain) were seeded at a density of 1.2×106 cells/ml in Dulbecco's modified Eagle's medium (DMEM; Gibco, Carlsbad, CA, USA) containing 10% FBS (Hyclone, Logan, UT, USA) and 1% penicillin-streptomycin (Gibco, Carlsbad, CA, USA). Two weeks later, microglia were detached from flasks by mild shaking and filtered through a cell strainer (BD Falcon, Bedford, MA, USA) to remove astrocytes. After centrifugation (1,000×g) for 5 min, cells were resuspended in fresh DMEM containing 10% FBS and 1% penicillin-streptomycin and plated at a final density of 1.5×105 cells/well on a 24 multiwell culture plate. After 2 hrs, the medium was changed for DMEM containing 5% FBS and 500 µM B27 supplement (Gibco, Carlsbad, CA, USA).
Primary microglial cells were treated with serum-free DMEM containing 100 ng/ml of LPS (Sigma, St. Louis, MO, USA) for 18 hrs. After washing with twice with DMEM, medium was replaced with fresh DMEM. LPS-conditioned media (LCM) was collected 24 hrs later and concentrated from 600 µl to 20 µl using a Centricon 10 (Millipore, Billerica, MA, USA). For control, primary microglial cells were treated with serum-free DMEM for 24 hrs.
Primary microglia cells (1.5×105) were seeded in 24-well plates and 1 day later treated with LCM or LPS (100 ng/ml). To measure the amount of NO produced, 50 µl of conditioned medium was mixed with an equal volume of Griess reagent (0.5% sulfanilamide, 0.05% N-naphthylene-diamine-H-chloride, and 2.5% H3PO4) and incubated for 5 min at room temperature. Absorbances of mixtures were measured at 550 nm using a microplate reader. NaNO2 standards were used to calculate NO2- concentrations.
Cells were seeded at a density of 1.5×105 cells per well in 24-well plates. All transfections were performed using oligofectamine (Invitrogen, Carlsbad, CA, USA) as a carrier. SMART pool siRNA specifically targeting HMGB1 (ON-TARGET plus SMART pool siRNA L-114889, accession no.NM_ 001109373; Dharmacon, Lafayette, CO, USA) and non-specific siRNA (on-TARGET plus Non-targeting pool D-001810, Dharmacon, Lafayette, CO, USA) were used. Transient transfections were carried out according to the manufacturer's instruction. siRNA and lipid complexes were added to the wells of 96-well culture plates to a final concentration of 40 pM siRNA and 1.8 µl oligofectamine.
Microglial cells were treated with LCM or LPS (100 ng/ml) and total RNA was purified with TRI reagent (Sigma-Aldrich, St Louis, MO) according to the manufacturer's instructions. First-strand cDNA was synthesized using the Takara RNA PCR kit (Takara Bio, Otsu, Japan) in a total volume of 20 µl containing 1 µg of total RNA. Real-time PCR was performed in a final volume of 20 µl containing 10 µl of 2x SYBR Green supermix (Takara Bio, Otsu, Japan), 1 µl each of 5 pmol/µl forward and reverse primers, and 5 µl of cDNA (50 ng; 1/100 dilution) using the Mini-Opticon Real-Time PCR System Detector (Bio-Rad, Richmond, CA). PCR was performed using: 5 minutes at 95℃, followed by 40 cycles of 30 seconds at 95℃, 30 seconds at 57℃, and 30 seconds at 72℃. The specificity of amplification was determined by DNA melting curve analysis using the built-in software. Differences in amplification fold were calculated based on the real-time PCR amplification of the target gene versus GAPDH using the built-in Gene Expression Analysis software in an iCycler iQ Real-Time RCR Detection System (Bio-Rad, Richmond, CA). The following primers sets were used; 5'-TCATTGAC CTCAACTACATGGT-3' and 5'-CTAAGCAGTTGGTGGTGCAG-3' for GAPDH. Real-Time PCR was performed in quadruplicate.
To analyze the binding of HBHP to HMGB1, a pull-down assay was performed using a 20 µl of streptavidin agarose beads (50% slurry) (Pierce, Rockford, IL, USA). Pull-down complexes were separated by 12% SDS-PAGE and analyzed by Western blotting. Biotin-labeled HBHP (HMSKPVQ; 1 or 5 µg/ml) or scrambled HBHP (PMQSKHV; 5 µg/ml) was incubated with LCM (containing HMGB1) at 4℃ for 4 hrs with rotation. A competition assay was performed using 5 µg/ml of HBHP, which was preincubated with the A box domain (1, 5, or 10 µg/ml) or the B box domain (1, 5, or 10 µg/ml) of HMGB1 for 1 hr at 4℃ with rotation. The biotin complexes obtained were incubated with streptavidin beads for 1 hr at 4℃ with rotation, centrifuged at 8,000 rpm for 1 min, and analyzed by Western blotting.
Cells were washed twice with cold PBS and lysed with RIPA buffer (50 mM Tris-HCl (pH 7.4), 1% NP40, 0.25% sodium-deoxycholate, 150 mM NaCl) containing 1 complete Mini protease inhibitor cocktail tablet (Roche diagnostics, Basel, Switzerland). For protein preparations from the supernatant, media were collected and concentrated as previously described and cells were treated with RIPA buffer. The lysates obtained were incubated in ice for 15 min and centrifuged at 12,000 rpm for 15 min at 4℃ and supernatants were loaded onto 12% SDS-PAGE gels. The primary antibodies (diluted at 1:1,000) used were as follows: anti-HMGB1 (Abcam, Cambridge, UK) and anti-α-tubulin (Santa Cruz Biotechnology, Santa Cruz, CA, USA). Primary antibodies were detected using a chemiluminescence kit (Roche, Mannheim, Germany) using goat anti-rabbit or anti-mouse horseradish peroxidase-conjugated secondary antibody (1:2,000, Millipore, Billerica, MA, USA).
Statistical analysis was performed by analysis of variance (ANOVA) followed by the Newman-Keuls test. All data are presented as means±SEMs and statistical significance was accepted at the 5% level.
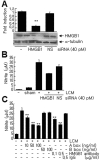
HMGB1 levels in primary microglia culture media were examined at after 1, 3, 6, 12, 18, or 24 hrs of LPS treatment (100 ng/ml). HMGB1 accumulation was detected after 3 hrs of LPS treatment and its levels gradually increased to peak at 18 hrs (Fig. 1A). On the other hand, HMGB1 levels in whole cell lysates decreased gradually from 3 hrs (Fig. 1B), whereas α-tubulin levels were similar at all time points (Fig. 1B).
To obtain LPS-free LCM, culture media were replaced with fresh medium after 18 hrs of LPS treatment and LCM was collected at 12 or 24 hrs later. Immunoblot analysis revealed HMGB1 accumulation in LPS-free LCM, and the level of HMGB1 at 24 hrs after medium replacement was slightly higher than that at 12 hrs (Fig. 1C). When fresh primary microglia cultures were incubated with LCM for 24 hrs, NO was induced (Fig. 1D) and more NO was induced when cells were treated with concentrated LCM (Fig. 1D). In following experiments, we used LCM collected 24 hrs after medium replacement that was concentrated two-fold.
To determine whether HMGB1 in LCM was responsible for microglial activation, the proinflammatory potency of LCM was examined after HMGB1 knockdown. HMGB1 levels in primary microglia decreased to 28.6±3.9% of the control level at 12 hrs after HMGB1-siRNA transfection (Fig. 2A). Furthermore, the LCM of HMGB1-siRNA transfected cells induced significantly less NO (59.9±0.9%) than LCM-treated control cells (Fig. 2B). However, NO levels were unchanged, when LCM from non-specific siRNA (NS)-transfected cells was used (Fig. 2B). These results suggest that HMGB1 plays an important role in LCM-induced microglial activation. LCM-induced NO production was also significantly suppressed by co-treating cells with HMGB1 A box (50 ng/ml) to 46.2±2.3% (n=4, p<0.01), a well-known antagonist of HMGB1 [20], or by co-treating anti-HMGB1 antibody (500 ng/ml) to 50.5±3.3% (n=4, p<0.01). These findings further support the critical role played by HMGB1 in LCM-induced microglia activation (Fig. 2C).
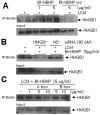
To determine whether HBHP affects LCM-induced microglia activation, NO levels were measured after co-treating cells with LCM and HBHP. NO levels were suppressed to 56.7±5.1%, 30.8±1.6%, and 32.5±10.8% of the control when HBHP was treated at 10, 50, or 100 ng/ml, respectively (Fig. 3A). Whereas scrambled HBHP (PMQSKHV; sc-HBHP, 50 or 100 ng/ml) did not produce such effects. In addition, proinflammatory cytokine productions in LCM-treated primary microglia cultures were also suppressed by HBHP treatment (10, 50, or 100 ng/ml), but not by scrambled HBHP (sc-HBHP, 50 or 100 ng/ml) (Fig. 3B), further supporting the notion that HBHP has anti-inflammatory effects in LCM-treated primary microglia cultures.
Next, we examined whether HBHP binds to the HMGB1 released by activated microglia to LCM. A biotin pull-down assay using biotin-labeled HBHP revealed the presence of binding between HBHP and HMGB1 in LCM in a HBHP-dose dependent manner, whereas, this binding was not detected when scrambled HBHP was used (Fig. 4A). Moreover, the formation of HBHP-HMGB1 was significantly suppressed when the LCM of HMGB1-ablated cells was used (Fig. 4B). When LCM was incubated with biotinylated HBHP (5 µg/ml) in the presence of HMGB1 A box (1, 5, or 10 µg/ml) or B box (5 or 10 µg/m), HMGB1/HBHP binding was clearly inhibited dose dependently by HMGB1 A box but not by HMGB1 B box (Fig. 4C). Accordingly, these results suggest that HBHP binds directly and specifically with HMGB1 A box in LCM and that the suppression of LCM-induced microglial activation by HBHP is due to this binding.
The present study shows that HMGB1 rapidly accumulates in LPS-treated primary microglia culture media (LCM), and that LCM is capable of activating microglia, and thus, of inducing proinflammatory markers. Furthermore, the study provides evidence that HBHP suppresses LCM-induced microglial activation, and that this is achieved by its direct binding to HMGB1 A box. In a previous report, we found that intranasal delivered HBHP ameliorates neuronal damage in the postischemic rat brain, and that HBHP confers neuroprotection to NMDA- or Zn2+-treated primary cortical cultures, wherein HBHP interacts with accumulated HMGB1 in NMDA-conditioned media [19]. Under pathological conditions, including that of the postischemic brain, where HMGB1 might be released from neurons after an excitotoxic insult [3] or secreted from activated microglia (Figs. 1 and 4), the neuroprotective effect of HBHP might be associated with its direct targeting of HMGB1 from various cellular origins. In view of the findings that HBHP has anti-inflammatory (present study) and neuroprotective effects [19], the robust neuroprotective effect of HBHP in the postischemic brain [19] is due to the combination of its anti-excitotoxic and anti-inflammatory effects.
HBHP suppressed LCM-induced nitrite production in HMGB1-ablated primary microglial cultures to 59.9±0.9% and suppressions of LCM-induced nitrite production in A box- or HMGB1 antibody-treated cells were 46.2±2.3% and 50.5±3.3%, respectively. These results indicate that although HMGB1 plays a crucial role in inflammation, factors localized in LCM other than HMGB1 might also play a role. Regarding binding region(s) on HMGB1, competition with recombinant HMGB1 A box in a pull-down experiment revealed that HBHP probably interacts with HMGB1 A box (Fig. 4C), which concurs with our previous result that was obtained under cell free conditions [19]. However, we do not exclude the possibility that different sites of HMGB1 are also involved in HBHP-HMGB1 binding and that site involvements depend on the cell and/or activating stimulus types, since relationship between differential modifications of HMGB1 and its specific function has been reported [21, 22] and these modifications might affect the interaction with HBHP.
Accumulating evidence indicate that HMGB1 forms complexes with various exogenous or endogenous molecules. For example, HMGB1 binds to LPS, IL-1β, and ssDNA, and the resulting complexes act as inflammatory inducers or enhancers [23-25]. In addition, an interaction between HMGB1 and integrin αvβ3 and phosphatidyl serine has been reported to be involved in the modulation of macrophage phagocytic activity [26, 27]. Interactions between HMGB1 and LPS, IL-1β, integrin αvβ3, and phosphatidylserine usually occur in the extracellular space, after passive HMGB1 release or active secretion. Furthermore, HMGB1 might bind to cytoplasmic proteins, such as, Beclin and src kinase [28, 29]. Here, we speculate that HBHP might interact with extracellular HMGB1 and this interaction might affect HMGB1 interactions with above-mentioned extracellular partners. However, we do not exclude the possibility that HBHP penetrates the cell membrane and interacts with intracellular HMGB1.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
- Scaffidi P, Misteli T, Bianchi ME. Release of chromatin protein HMGB1 by necrotic cells triggers inflammation. Nature 2002;418:191-195.

- Bonaldi T, Talamo F, Scaffidi P, Ferrera D, Porto A, Bachi A, Rubartelli A, Agresti A, Bianchi ME. Monocytic cells hyperacetylate chromatin protein HMGB1 to redirect it towards secretion. EMBO J 2003;22:5551-5560.
- Kim JB, Sig Choi J, Yu YM, Nam K, Piao CS, Kim SW, Lee MH, Han PL, Park JS, Lee JK. HMGB1, a novel cytokine-like mediator linking acute neuronal death and delayed neuroinflammation in the postischemic brain. J Neurosci 2006;26:6413-6421.
- Bianchi ME, Manfredi AA. High-mobility group box 1 (HMGB1) protein at the crossroads between innate and adaptive immunity. Immunol Rev 2007;220:35-46.
- Andersson U, Wang H, Palmblad K, Aveberger AC, Bloom O, Erlandsson-Harris H, Janson A, Kokkola R, Zhang M, Yang H, Tracey KJ. High mobility group 1 protein (HMG-1) stimulates proinflammatory cytokine synthesis in human monocytes. J Exp Med 2000;192:565-570.
- Dumitriu IE, Baruah P, Valentinis B, Voll RE, Herrmann M, Nawroth PP, Arnold B, Bianchi ME, Manfredi AA, Rovere-Querini P. Release of high mobility group box 1 by dendritic cells controls T cell activation via the receptor for advanced glycation end products. J Immunol 2005;174:7506-7515.
- Gibot S, Massin F, Cravoisy A, Barraud D, Nace L, Levy B, Bollaert PE. High-mobility group box 1 protein plasma concentrations during septic shock. Intensive Care Med 2007;33:1347-1353.
- Wang H, Bloom O, Zhang M, Vishnubhakat JM, Ombrellino M, Che J, Frazier A, Yang H, Ivanova S, Borovikova L, Manogue KR, Faist E, Abraham E, Andersson J, Andersson U, Molina PE, Abumrad NN, Sama A, Tracey KJ. HMG-1 as a late mediator of endotoxin lethality in mice. Science 1999;285:248-251.
- Kocsis AK, Szabolcs A, Hofner P, Takács T, Farkas G, Boda K, Mándi Y. Plasma concentrations of high-mobility group box protein 1, soluble receptor for advanced glycation end-products and circulating DNA in patients with acute pancreatitis. Pancreatology 2009;9:383-391.
- Goldstein RS, Gallowitsch-Puerta M, Yang L, Rosas-Ballina M, Huston JM, Czura CJ, Lee DC, Ward MF, Bruchfeld AN, Wang H, Lesser ML, Church AL, Litroff AH, Sama AE, Tracey KJ. Elevated high-mobility group box 1 levels in patients with cerebral and myocardial ischemia. Shock 2006;25:571-574.
- Agnello D, Wang H, Yang H, Tracey KJ, Ghezzi P. HMGB-1, a DNA-binding protein with cytokine activity, induces brain TNF and IL-6 production, and mediates anorexia and taste aversion. Cytokine 2002;18:231-236.
- Kim SW, Lim CM, Kim JB, Shin JH, Lee S, Lee M, Lee JK. Extracellular HMGB1 released by NMDA treatment confers neuronal apoptosis via RAGE-p38 MAPK/ERK signaling pathway. Neurotox Res 2011;20:159-169.
- Liu K, Mori S, Takahashi HK, Tomono Y, Wake H, Kanke T, Sato Y, Hiraga N, Adachi N, Yoshino T, Nishibori M. Anti-high mobility group box 1 monoclonal antibody ameliorates brain infarction induced by transient ischemia in rats. FASEB J 2007;21:3904-3916.
- Zhang J, Takahashi HK, Liu K, Wake H, Liu R, Maruo T, Date I, Yoshino T, Ohtsuka A, Mori S, Nishibori M. Anti-high mobility group box-1 monoclonal antibody protects the blood-brain barrier from ischemia-induced disruption in rats. Stroke 2011;42:1420-1428.
- Jin YC, Kim SW, Cheng F, Shin JH, Park JK, Lee S, Lee JE, Han PL, Lee M, Kim KK, Choi H, Lee JK. The effect of biodegradable gelatin microspheres on the neuroprotective effects of high mobility group box 1 A box in the postischemic brain. Biomaterials 2011;32:899-908.
- Kim SW, Jin Y, Shin JH, Kim ID, Lee HK, Park S, Han PL, Lee JK. Glycyrrhizic acid affords robust neuroprotection in the postischemic brain via anti-inflammatory effect by inhibiting HMGB1 phosphorylation and secretion. Neurobiol Dis 2012;46:147-156.
- Kim ID, Shin JH, Kim SW, Choi S, Ahn J, Han PL, Park JS, Lee JK. Intranasal delivery of HMGB1 siRNA confers target gene knockdown and robust neuroprotection in the postischemic brain. Mol Ther 2012;20:829-839.
- Dintilhac A, Bernués J. HMGB1 interacts with many apparently unrelated proteins by recognizing short amino acid sequences. J Biol Chem 2002;277:7021-7028.
- Kim ID, Shin JH, Lee HK, Jin YC, Lee JK. Intranasal delivery of HMGB1-binding heptamer peptide confers a robust neuroprotection in the postischemic brain. Neurosci Lett 2012;525:179-183.
- Yang H, Ochani M, Li J, Qiang X, Tanovic M, Harris HE, Susarla SM, Ulloa L, Wang H, DiRaimo R, Czura CJ, Wang H, Roth J, Warren HS, Fink MP, Fenton MJ, Andersson U, Tracey KJ. Reversing established sepsis with antagonists of endogenous high-mobility group box 1. Proc Natl Acad Sci U S A 2004;101:296-301.
- Antoine DJ, Williams DP, Kipar A, Laverty H, Park BK. Diet restriction inhibits apoptosis and HMGB1 oxidation and promotes inflammatory cell recruitment during acetaminophen hepatotoxicity. Mol Med 2010;16:479-490.
- Kazama H, Ricci JE, Herndon JM, Hoppe G, Green DR, Ferguson TA. Induction of immunological tolerance by apoptotic cells requires caspase-dependent oxidation of high-mobility group box-1 protein. Immunity 2008;29:21-32.
- Youn JH, Oh YJ, Kim ES, Choi JE, Shin JS. High mobility group box 1 protein binding to lipopolysaccharide facilitates transfer of lipopolysaccharide to CD14 and enhances lipopolysaccharide-mediated TNF-alpha production in human monocytes. J Immunol 2008;180:5067-5074.
- Sha Y, Zmijewski J, Xu Z, Abraham E. HMGB1 develops enhanced proinflammatory activity by binding to cytokines. J Immunol 2008;180:2531-2537.
- Ivanov S, Dragoi AM, Wang X, Dallacosta C, Louten J, Musco G, Sitia G, Yap GS, Wan Y, Biron CA, Bianchi ME, Wang H, Chu WM. A novel role for HMGB1 in TLR9-mediated inflammatory responses to CpG-DNA. Blood 2007;110:1970-1981.
- Friggeri A, Yang Y, Banerjee S, Park YJ, Liu G, Abraham E. HMGB1 inhibits macrophage activity in efferocytosis through binding to the alphavbeta3-integrin. Am J Physiol Cell Physiol 2010;299:C1267-C1276.
- Liu G, Wang J, Park YJ, Tsuruta Y, Lorne EF, Zhao X, Abraham E. High mobility group protein-1 inhibits phagocytosis of apoptotic neutrophils through binding to phosphatidylserine. J Immunol 2008;181:4240-4246.
- Kang R, Livesey KM, Zeh HJ, Loze MT, Tang D. HMGB1: a novel Beclin 1-binding protein active in autophagy. Autophagy 2010;6:1209-1211.
- Banerjee S, de Freitas A, Friggeri A, Zmijewski JW, Liu G, Abraham E. Intracellular HMGB1 negatively regulates efferocytosis. J Immunol 2011;187:4686-4694.