Articles

Article Tools
 View Full Text View Full Text |
 Abstract Abstract |
 Article as PDF Article as PDF |
 Print this Article Print this Article |
 Pubmed Pubmed |
 PMC PMC |
 PubReader PubReader |
 Export to Citation Export to Citation |
 Email Alerts Email Alerts |
 Open Access Open Access |
Share this article on :



Stats or Metrics
Article
Original Article
Exp Neurobiol 2020; 29(1): 93-105
Published online February 29, 2020
https://doi.org/10.5607/en.2020.29.1.93
© The Korean Society for Brain and Neural Sciences
BMD42-2910, a Novel Benzoxazole Derivative, Shows a Potent Anti-prion Activity and Prolongs the Mean Survival in an Animal Model of Prion Disease
Jae Wook Hyeon1, Ran Noh1, Jiwon Choi2, Sol Moe Lee1, Yeong Seon Lee1, Seong Soo A. An3, Kyoung Tai No2,4 and Jeongmin Lee5*
1Division of Bacterial Disease Research, Center for Infectious Disease Research, Korea National Institute of Health, Centers for Disease Control and Prevention, Cheongju 28160, 2Bioinformatics and Molecular Design Research Center, Yonsei University, Seoul 03722, 3Gachon Bio Nano Research Institute, Gachon University, Seongnam 13120, 4Department of Biotechnology, College of Life Science and Biotechnology, Yonsei University, Seoul 03722, 5Division of Research Planning, Korea National Institute of Health, Centers for Disease Control and Prevention, Cheongju 28160, Korea
Correspondence to: *To whom correspondence should be addressed.
TEL: 82-43-719-8024, FAX: 82-43-719-8489
e-mail: jeongminlee@korea.kr
This is an Open Access article distributed under the terms of the Creative Commons Attribution Non-Commercial License
(http://creativecommons.org/licenses/by-nc/4.0) which permits unrestricted non-commercial use, distribution, and
reproduction in any medium, provided the original work is properly cited.
Abstract
Prion diseases are a group of neurodegenerative and fatal central nervous system disorders. The pathogenic mechanism involves the conversion of cellular prion protein (PrPC) to an altered scrapie isoform (PrPSc), which accumulates in amyloid deposits in the brain. However, no therapeutic drugs have demonstrated efficacy in clinical trials. We previously reported that BMD42-29, a synthetic compound discovered
Graphical Abstract
Keywords: Transmissible spongiform encephalopathy, Prion protein, Anti-prion, Derivatives, In silico
INTRODUCTION
Prion diseases, or transmissible spongiform encephalopathies, are a group of neurodegenerative diseases, which share similar pathogenic mechanisms; the prion protein (PrP) undergoes posttranslational modification and is converted from an endogenous (PrPC) to a pathogenic isoform (PrPSc) [1–3]. This conversion leads to the changes of chemical and physical properties, including insolubility in nondenaturing detergents, partial resistance to protease digestion, and a high tendency to form aggregates and amyloid fibrils [4–6]. Prion diseases are associated with PrPSc accumulation in the brain, specifically of PrP amyloid fibrils. Currently, the mechanisms underlying this conversion are incompletely defined [7, 8]. The invariable lethality of prion diseases necessitates the identification of compounds that prevent misfolding of PrPC, replication, or accumulation of PrPSc [9, 10].
Current knowledge on prion diseases has revealed potential therapeutic strategies, including the prevention of the conversion to PrP aggregates, induction of degradation of these aggregates, destabilization the PrPSc structure, or interference with PrPC/PrPSc cellular uptake [11]. Therapeutic trials have been conducted using a wide variety of potential prion diseases treatments, and numerous compounds have been extensively investigated to demonstrate antiprion activity in cell culture models [12–14]. However, no current treatment has sufficient activity to halt disease progression in infected animal models. The therapeutic use of these compounds is restricted by their intrinsic cytotoxicity and pharmacokinetic properties of drug delivery, including absorption and circulation, as well as by their limited ability to pass through the bloodbrain barrier (BBB), resulting in failed efficacy
We previously described the discovery of small antiprion molecules obtained from a combined structure and ligandbased virtual screening system
MATERIALS AND METHODS
Compounds
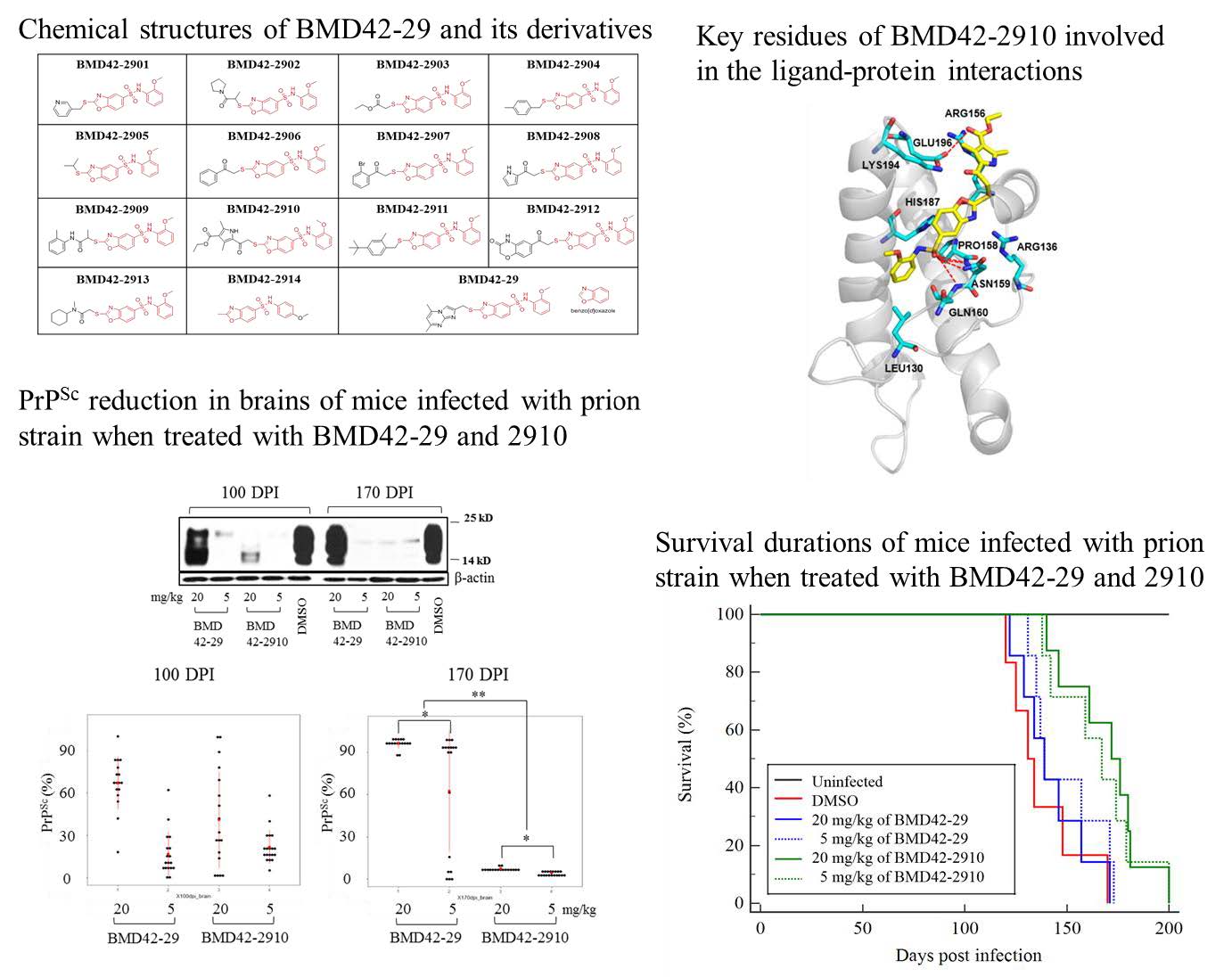
The derivatives having a core structure of BMD4229 (Nphenylbenzo[ d]oxazole5sulfonamide) were designed and docked into the 2D structure of PrPC using the ChemDraw Ultra as reported before [25]. As the final outcome, 14 derivatives with high binding energy were selected, named as BMD422901 to BMD422914, and their structures were optimized. BMD422901 to 2909 were synthesized in the Enamine (Monmouth Junction, NJ, USA), BMD422910 to 2912 were synthesized in the Akos Consulting & Solutions GmbH (Steinen, Germany) and BMD422913 and BMD422914 were synthesized in the Interchem (Paramus, NJ, USA) and ChemDiv(San Diego, CA, USA), respectively. For
Inhibition of PrPres in scrapieinfected cells
Prioninfected murine neuroblastoma (ScN2a) cells, derived from the neuroblastoma2a cell line of the American Type Culture Collection are stably infected with PrPSc (Rocky Mountain Laboratory [RML] strain); the cells were generously provided by Dr. Ryu at Hanyang University in Republic of Korea. Cell culture was performed as described previously [25]. Briefly, cells were seeded in T25 cm2 flasks (TPP, Z707481) at 2×105 cells, and preincubated in DMEM (Gibco, Big Carbin, OK, USA) (12430) containing 10% fetal bovine serum, 1% penicillinstreptomycin (Gibco, 15140), and 2 mM Lglutamine (Gibco, 25030) for 24 h under 5% CO2 at 37°C. The media were replaced with fresh media containing the test compounds. After incubation for 3 days, the media were again replaced with fresh media containing the test compounds, and cells were incubated for 3 days more. Finally, the cells were harvested after incubation with the compounds for 6 days. The stock solutions of 14 derivatives were diluted in DMEM to a final concentration of 20 μM. Control cell cultures were treated with DMSO only (0.1% v/v) and quinacrine (1 μM) as activity controls. Secondary cell testing was dosedependently performed at concentrations of 1.2, 2.5, 5, and 10 μM. Each compound was tested in duplicate, in three independent experiments.
The cells were rinsed once in PBS and then incubated with 0.1% (vol/vol) trypsinEDTA (Gibco, 25300) for 1 min at room temperature. The detached cells were centrifuged at 4000×g for 10 min at 4°C and rinsed once with PBS. Cells were lysed in icecold lysis buffer (10 mM EDTA, 10 mM Tris, pH 8.0, 100 mM NaCl, 0.5% [wt/vol] Nonidet P40, and 0.5% [wt/vol] sodium deoxycholate), with two freezethaw cycles in liquid nitrogen, and then sonicated at an amplitude of 30%. The total protein concentration of cell lysates was adjusted to 200 μg/mL.
Cell lysates were digested with PK (20 μg/mL) (Merck, Kenilworth, NJ, USA) (70663) for 60 min at 37°C. The reaction was stopped with 2 mM Pefabloc (Roche, 11429876001), and the lysates were centrifuged for 90 min at 20,000×g at 4°C. Pellets were resuspended in SDS sample buffer (125 mM TrisHCl, pH 6.8, 5% [vol/vol] glycerol, 6 mM EDTA, 5% [wt/vol] SDS, 0.04% [vol/vol] bromophenol blue, and 12.5% [vol/vol] βmercaptoethanol).
Protein mixtures were separated by SDSpolyacrylamide gel electrophoresis for 2 h at 150 V, at 4°C, using 12% gels. Proteins were then transferred to a PVDF membrane using the iBlot system (Invitrogen, Carlsbad, CA, USA) for 7 min. The membrane was blocked for 1 h at room temperature in 5% (wt/vol) skim milk in PBS containing 0.1% (vol/vol) Tween 20 (PBST), and then incubated overnight at 4°C with the monoclonal PrP antibody 6H4 (1:2500; Prionics, Zurich, Switzerland; PRN01011), diluted in 10% (vol/vol) blocking buffer. After washing with PBST, the membrane was incubated for 1 h at room temperature with HRPconjugated antimouse IgG (1:5,000; DAKO, Santa Clara, CA, USA; P0260). The signals were visualized using ECL (EBP1073; Elpis Biotech, Daejeon, Republic of Korea), and the protein bands were scanned using Image Scanner III (GE, Munich, Germany). The relative band densities are shown as the volume intensity/ mm2, relative to the βactin (1:5000; Cell Signaling technology, Beverly, MA, USA; 4970) band density.
Total PrP measurement
Total PrP in lysates of cells treated with desired compounds was measured using the ELISA kit for PRION (PRNP; Cloud-Clone Corp., Katy, Texas, USA) according to the manufacturer’s instructions. The absorbance at 450 nm was measured using a plate reader (VICTOR3, PerkinElmer, MA, USA). All samples were analyzed in triplicate.
Inhibition of PrPres in RTQuIC
Human recombinant PrP (rPrP, residues 23–231; accession no. K02234) was produced, and the RTQuIC reaction was performed as described in our previous report, as a modified method of Wilham et al. and Cramm et al. [28–30]. Briefly, Escherichia coli transfected with vector with human PrP sequence was grown in Luria broth medium with kanamycin and chloramphenicol. Protein was expressed by Overnight Express Autoinduction system (Novagen, Darmstadt, Germany; 71300). The rPrP was purified from inclusion bodies using NiNTA Superflow resin (Qiagen, Hilden, Germany; 30450). The protein was refolded under gradient condition, eluted with 10 mM phosphate buffer (pH 5.8). Protein purity was measured by SDSPAGE, mass spectrometry. The final composition of the reaction buffer was as follows: 162 mM phosphate buffer (pH 6.9), 170 mM NaCl, 1 mM EDTA, 10 μM Thioflavin T (ThT), and 0.1 mg rPrP. Ninetysix microliters of buffer were dispensed into each well of a clearbottomed black 96well microplate (Nunc, NY, USA; 265301).
Two microliters of diluted sporadic Creutzfeldt Jacob disease (sCJD) brain homogenate from the National Institute for Biological Standards and Control (NIBSC; reference code NHBX0/0001) were seeded into each well. Two microliters of each compound (BMD422908, 2909, 2910, and 2914; 1.2, 2.5, 5, 10, and 20 μM) and DMSO were added to designed wells. Compounds were solubilized in DMSO. The plate was sealed and incubated for 72 h at 44°C, with intermittent shaking using a BMG Labtech Optima Fluostar plate reader. Each reaction was performed in quadruplicate. A positive response was indicated if the mean of the two highest readings at 72 h was greater than twofold, compared to the negative control. In a previous study, to exclude the possibility that compounds were affecting the ThT fluorescence dye rather than substrate rPrP or PrP amyloid, RTQuIC products were collected from wells after the last reaction, and PrPres was detected using western blotting as mentioned above. Additionally, a dyeindependent conversion assay which is called Multimer Detection System (MDS) was performed to further confirm the nature of ThT. As a result, ThTcompound interference is not observed in optical (data not shown). This result was consistent with that of our previous experiment [30].
Inhibition of PrPres in mice
For
Brains and spleens of the mice euthanized at 100 and 170 DPI were removed, weighed, homogenized in buffer (PBS; pH 7.4) containing 1 mM EDTA, 5 M NaCl, Triton X100, and protease inhibitor, using a Precellys tissue homogenizer with glass beads to make 1% homogenate, and then used for western blotting. A total of 300 μL of plasma was prepared from 700 μL of whole blood, which was collected from the heart. From this sample, 30 μL of plasma mixed with 10 μL of sample buffer was used for western blotting. Mice that died from causes other than prion diseases were excluded from the data set [36]. The protein bands were scanned using Image Scanner III (GE). Scatter plots of individual data were generated using the ggplot2 software package with R software package (version 3.4.3).
Survival durations and hazardous factor analysis
Survival durations were analyzed by KaplanMeier survival analysis, and hazardous factors were then generated using MedCalc Statistical Software version 17.6 (MedCalc Software bvba, Ostend, Belgium;
Molecular docking
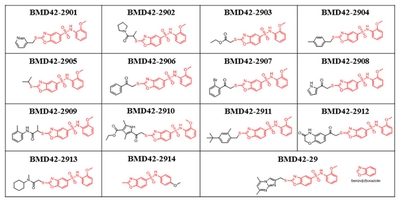
Molecular docking calculations were performed using the CDOCKER program to identify binding mode of novel anti-prion compound, which uses a CHARMm forcefield and grid-based molecular dynamics docking method. To determine the binding pose of compounds to the hotspot binding site of PrPC, the NMR structure of PrPC (1AG2) was retrieved from the Protein Data Bank. The hotspot regions of PrPC with key residues (Asn159, Gln160, Lys194, and Glu196) was used to define the binding site of PrPC. The best docking pose for each compound was selected based on the best scoring conformations from LigScore2, binding patterns, and visual inspection. The predicted structures of PrPC-compound complexes were visualized using the PyMOL program.
Statistical analyses
Each experiment was repeated three times. One-way analysis of variance with Tukey-Kramer post hoc tests was conducted using Microsoft Excel and SAS (version 9.3). Differences were considered statistically significant when p<0.05.
RESULTS
Derivatives
To investigate the interaction pattern of BMD42-29 on the hotspot regions of PrPC with key residues, we conducted molecular docking experiments. N-phenylbenzo[d]oxazole-5-sulfon-amide group of BMD42-29 interacted with Asn159 and Gln160 of PrPC and docked into a cavity surrounded by Pro 158, Asn159 and Gln160, which indicated that benzene sulfonamide group likely contributes to the tight binding with PrPC. Consequently, we decided to use N-phenylbenzo[d]oxazole-5-sulfonamide group of BMD42-29 as the starting point for further compound modifications. From the interaction patterns of the hotspot region, we expected that the substitution of a imidazo[1,2-α]pyrimidine group would allow compounds with other substituents at this position to bind the PrPC and increase the binding specificity. In order to identify BMD42-29 derivatives of the hotspot region binding interaction with core scaffolds, we performed substructure search screening from integrated database. Finally, the 14 derivatives were identified by docking simulations and evaluated for experimental validation.
Antiprion activity of derivatives in a cell model
Inhibition of PrPres amplification was observed in ScN2a cells treated with 14 derivatives (Fig. 2). As previously reported, no cytotoxicity was observed in compounds derived
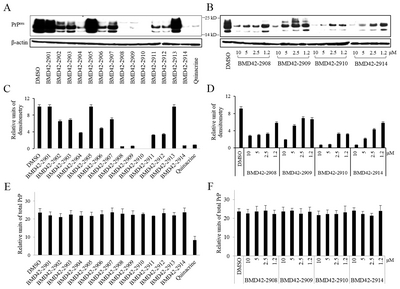
The four effective derivatives reduced PrPres levels even at 1.2 μM. The IC50 values were 1.8 μM for BMD422908, 5 μM for BMD422909, <1.2 μM for BMD422910, and 2.1 μM for BMD422914. More than 90% inhibition was observed at 5 and 10 μM BMD422910 and at 10 μM BMD422914.Given doses of the derivatives with similar activity, BMD422910 was the most effective derivative compound. Semiquantitative intensities for Fig. 2A and 2B bands are provided in Fig. 2C and 2D, respectively. The expression of total PrP was constant in conditions of various compounds and doses, except for quinacrine (Fig. 2E and 2F). This suggested that BMD422910 as well as other derivatives only affected PrPres at low concentrations without change of total PrP.
Antiprion activity of derivatives in RTQuIC
Blocking activity of PrPC conversion was observed by RTQuIC using BMD422908, BMD422909, BMD422910, and BMD422914 with the same doses (Fig. 3). Increased and decreased fluorescence indicate that PrP aggregation is generated and blocked, respectively. The fluorescence was increased in DMSO controls of all experiments at 5~11 h, indicating that PrPC conversion was rapidly generated without any disruptions. In case of BMD422908, fluorescence is not elevated at 20 μM until 62 h, whereas it was elevated in most concentrations at 18~22 h, indicating that there was no blocking activity at most doses of BMD422908. Also, the higher the dose, the later the amplification began, indicating that showed the partial blocking activity dosedependently (Fig. 3A).
In case of BMD422909, fluorescence was increased at 8~18 h in all doses similar to that of the DMSO control, indicating that PrPC conversion was not blocked by BMD422909 and even it did not show the partial blocking activity (Fig. 3B). In case of BMD422910, the baseline level of fluorescence was maintained at 5, 10, and 20 μM until the last reading (72 h), except that the fluorescence was increased at around 62 h in wells treated with 1.2 and 2.5 μM (Fig. 3C). This result suggested that PrPC conversion was completely blocked until the last reaction, and also the concentration required for blocking activity was relatively low in BMD422910. In case of BMD422914, the baseline fluorescence was maintained at 10 and 20 μM until the last reading (72 h), except that the fluorescence was partially elevated at 1.2, 2.5, and 5 μM (Fig. 3D). Given that the threshold concentration of BMD422910 showing completely blocking activity was 5 μM and the partial blocking activity was also much more effective than that of other derivatives, BMD422910 was the most effective antiprion activity.
Toxicological assessments of BMD42-29 and BMD42-2910 in uninfected mice
Based on cell and RT-QuIC assays, BMD42-2910 was selected for further
Antiprion activity of BMD4229 and BMD422910 in PrPScinfected mice
Considering both the optimal inhibitory concentration of 20 μM (10 mg/kg) in cells and RTQuIC and the maximum of 100 mg/kg in
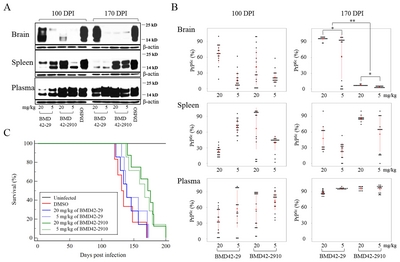
Each individual immunoblot data were displayed as scatter plots (Fig. 4B). There were individual differences in all brains at 100 DPI. However, most data were distributed without individual differences in the brains at 170 DPI, except for 5 mg/kg of BMD4229 at 170 DPI. Although PrPres of brain was reduced in 5 mg/kg BMD4229, 5 mg/kg BMD422910, and 20 mg/kg BMD422910 at 100 DPI as shown in Fig. 4A, the individual differences may not support the conclusion that they represented antiprion activity. Therefore, BMD4229 and BMD422910 partially reduced PrPres of brain at 100 DPI. By contrast, PrPres of brain was almost completely reduced in 5 and 20 mg/kg BMD422910 at 170 DPI. It was consistent with their individual data without the differences. Hence, the findings in brains that 5 and 20 mg/kg of BMD422910 showed antiprion activity at 170 DPI were reliable. As was the case in brains, the individual differences of PrPres of pleen was hard to conclude that they represented antiprion activity. It was consistently ineffective in plasma. But in part, the antiprion activity was higher in spleen than in plasma, considering degree of variation between individuals.
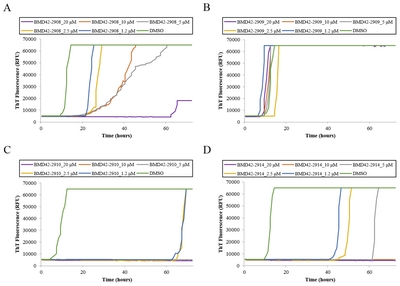
Furthermore, survival rates were measured in PrPScinfected mice treated with BMD4229 and BMD422910. KaplanMeier survival analysis demonstrated that mice treated with 5 and 20 mg/kg BMD422910 survived longer than mice treated with 5 and 20 mg/kg BMD4229 or DMSO (Fig. 4C). Survival durations were increased by approximately 7 and 30 days in mice treated with BMD4229 and BMD422910, respectively. Mean hazard ratios with 95% confidence intervals indicated that it was reduced in treatment of BMD4229 and BMD422910, as compared with that of DMSO (Table 2).
The docking conformation of BMD4229 and BMD422910 to PrPC at specific amino acids
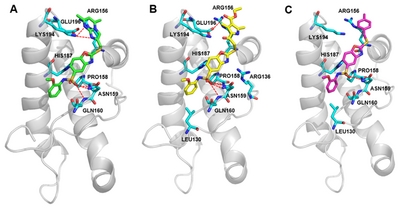
The predicted binding modes of BMD4229, BMD422910, and BMD422904 on PrPC hotspot binding site were shown in Fig. 5. We identified that the docked pose of BMD422910 has a similar conformation to that of PrPCBMD4229 complexes. After applying the LigScore2 scoring functions for filtering, the best poses were selected based on exhibiting good interactions with hotspot residues (Asn159, Gln160, Lys194, and Glu196). The BMD4229 and BMD422910 interacted strongly with Asn159, Gln160 and Lys194 or Glu196 by forming hydrogen bonding interactions, as well as intermolecular πstacking interactions through its benzoxazole ring (Fig. 5A and 4B). However, BMD422904 failed to forms hydrogen bonds with Lys194 or Glu196 because of its relatively smaller substituted ring size (Fig. 5C). These results demonstrated the central role of Asn159, Gln160, Lys194 and Glu196 in the binding affinity of antiprion compounds to PrPC. These interaction patterns of BMD422910 suggested that possible chemical modifications at the top hydrophobic pocket can lead to increase the binding affinity of the compounds. The remaining active compounds did not show binding scores high enough to allow the simulation of their docking to PrPC.
DISCUSSION
Existing antiprion compounds include those described in library databases, FDA approved drugs for treating other diseases, natural products, antibodies, peptides and derivatives [12]. The inhibitory effects of these compounds on PrPres are reliably identified in PrPScinfected cell models; however, therapeutic effects are not often observed in PrPScinfected mice, and clinical trials have been unsuccessful to date [13]. An advantage of exploring antiprion compounds
In a previous study, BMD4229 and BMD4235 were the most effective antiprion compounds tested [25]. BMD4229 displayed optimal properties, exhibiting inhibition of PrPres amplification and stronger binding affinity than other previously reported antiprion compounds [40, 41]. However, it did not produce a marked reduction in PrPC levels, possibly indicating that it stabilizes PrPC and inhibits its pathological conformational change to PrPres. In contrast, BMD4235 produced the highest antiprion activity in both PrPres and PrPC levels, although it had lower binding affinity, which suggests that BMD4235 acts by either stabilizing or eliminating PrPC and may control PrPC via nonspecific mechanisms. Hence, we found the derivatives of BMD4229 based on its remarkable inhibition for PrPres amplification and strong binding affinity, with the aim to determine more effective compounds via
Infectious forms of animal prions can be propagated in cell culture, notably in ScN2a cell lines [42, 43]. Various immunological methods are available to measure prion load in these cell lines, and therefore provide a valuable means of evaluating the antiprion activity of potential compounds. We performed RTQuIC by ThT fluorescence, as well as western blotting of cell lysates using a PrP antibody. RTQuIC is a recent innovation that allows realtime detection and monitoring of rPrP conversion to amyloid form [28]. RTQuIC has been successfully used to detect small amounts of PrPSc in brain tissue, cerebrospinal fluid, blood, saliva, and nasal fluid from human, cervid, ovine, bovine, and rodent species. PrP amyloid resulting from the conversion of rPrP to its aggregated form is indicated by increased ThT fluorescence, which directly indicates betasheet structure formation in protein aggregates [44].
Inhibitory effects of blocking PrPC conversion were confirmed both in our cell model and by RTQuIC
PrPres was dramatically reduced at 170 DPI in the brains of PrPScinfected mice treated with BMD422910, and this effect was sustained. Individual differences were not statistically significant; thus we could not conclude whether BMD4229 has antiprion activity
Administration of BMD422910 into the abdominal cavity is relatively convenient, requires a small amount of the compound, and has a higher absorption rate than oral administration because it bypasses the digestive system [45]. In our study, administration into the abdominal cavity resulted in high activity in the brain, as supported by several lines of evidence. First, the compound might be small enough to cross the BBB in order to directly affect PrPSc in the brain. Second, the activity of the compound could be markedly observed in brain areas with extensive PrPSc distribution. However, it was not easily distinguished in spleen or plasma, although the compound had suitable effects. This is likely because PrPSc in the spleen and plasma has relatively low infectivity and pathogenicity and is present at low concentration [46]. Therefore, high expression of PrPSc is likely necessary to generate the observed outcomes in the brain.
It has been proposed that the hydrogen bonds from strand S1 and helix B may prevent conversion of PrPC to PrPSc [47]. BMD4229 and BMD422910 had strong hydrogen bonds at Asn159, Gln160, Lys194 and Glu196 within PrPC and interacted with conserved PrPC hotspot residues, indicating the importance of hydrogen bonds interactions and the hydrophobic environment; this was predicted by the pharmacophore model. We found that BMD422904 shows partial antiprion activity in our PrPScinfected cell model, and also binds PrPC at Asn159 with a hydrogen bond. Although only one binding site was involved in the docking simulation, antiprion activity was feasible. Therefore, we infer that compound binding to Asn159 of PrPC may be effective. In a previous study, we reported that the hotspot regions in β1 (Asn159), α2 (Val189, Tyr192, and Lys194), and the α1~α2 loop (Glu196), which are involved in the pathogenic conversion process, are obstructed by binding to the antiprion compounds. In particular, the α1~α2 salt bridge between Arg156 and Glu196 of the misfolded PrPC returns to the PrPC state in the presence of BMD4229 [27]; this finding is consistent with other reports [39, 47]. BMD422910 has hydrogen bonding interactions at Glu196, which are not absent in BMD4229. Hence, the high antiprion activity of BMD422910 may result from binding to Glu196 of PrPC in the binding mode.
Future research should first assess whether PrPSc strains differ between cell (RML strain) and mouse (ME7 strain) models. As previously reported, prion studies often produce incompatible results, depending on which PrPSc strain is used [48]. Therefore, other PrPSc strains should be used in future studies, to enable generalization of BMD422910 activities. Second, BMD422910 should be tested using a more specific nonclinical trial in humanized models. For example, we plan to use PrPScinfected human neuronal cells and humanized transgenic mice to study potential humanhost effects. Third, cell signaling and cytokines should be analyzed in cells and in the brains of mice treated with BMD422910, to study the specific action of involved pathways. Finally, mutant PrPs at 159, 194 and 196 amino acid should be introduced in RTQuIC assays in order to determine if these positions are important for their binding or inhibition.
In conclusion, we discovered compounds that inhibit the conversion of PrPC to PrPres and validated the compounds using
ACKNOWLEDGEMENTS
This research was supported by a research grant [2014-NG52003-00] and funding from the Korea Centers for Disease Control and Prevention (4800-4838-303). RT-QuIC assay using sCJD brain homogenate and
Figures
{kind=link}
Chemical structures of BMD42-29 and its derivatives. The red components of the derivatives are templates originated from BMD42-29, and the black components are modifiable.
{kind=link}
Inhibition of PrPres amplification in ScN2a cells treated with 14 derivatives of BMD42-29. Effects of 14 derivatives (20 μM) (A), dose-dependent four effective derivatives (1.2~10 μM) (B) on inhibition of PrPres amplification in ScN2a cells were assessed by western blotting. Relative units of densitometry for (A) and (B) indicate the volume intensity/mm2, relative to β-actin signal in (C) and (D), respectively. Total PrP was measured in ScN2a cells treated with 14 derivatives (20 μM) (E) and dose-dependent four effective derivatives (1.2~10 μM) (F). Controls were treated with DMSO and quinacrine. Each value represents the mean±standard deviation; p<0.05. Three independent experiments were performed in duplicate, and representative immunoblots are shown. Molecular mass markers are indicated to the between two blots.
{kind=link}
Blocking activity of four derivatives against
{kind=link}
Inhibition of PrPres amplification in ME7-infected mice treated with BMD42-29 or BMD42-2910. Effects of BMD42-29 and BMD42-2910 on PrPres amplification in ME7-infected mice were analyzed. The infected mice were treated with 5 and 20 mg/kg of the compounds and DMSO. At 100 and 170 DPI, seventeen to nineteen mice per a group were euthanized, and their brain, spleen, and plasma were immunoblotted after PK treatment. Representative blots are shown per a group, with molecular mass markers indicated to the right of the blots (A). Individual PrPres levels were shown by scatter plots (B). Each spot corresponds to band intensity/DMSO band intensity ×100. *p<0.05. **p<0.001. Survival durations of the eight to ten mice per a group infected with ME7 and treated with 20 (line) and 5 (dotted line) mg/kg per week of BMD42-29 (blue), BMD42-2910 (green) or DMSO (red line), and uninfected mice treated with DMSO (black line) are shown by Kaplan-Meier curves (C).
{kind=link}
Molecular docking studies of the compounds with cellular prion protein (PrPC). Key residues (cyan sticks) involved in the ligand-protein interactions are shown as stick model and H-bonds are indicated by red dashed lines. The protein surface is colored in light gray and BMD42-29 (A), BMD42-2910 (B), and BMD42-2904 (C) are shown as green, yellow, and magenta sticks, respectively. Images were drawn with PyMol (Delano Scientific LLC, San Carlos, CA, USA).
Tables
Experimental design for
| Toxicity (n) | BMD42-29 | BMD42-2910 | DMSO | ||||
|---|---|---|---|---|---|---|---|
| 4 mg/kg | 20 mg/kg | 100 mg/kg | 4 mg/kg | 20 mg/kg | 100 mg/kg | ||
| Male | 5 | 5 | 5 | 5 | 5 | 5 | 5 |
| Female | 5 | 5 | 5 | 5 | 5 | 5 | 5 |
| Female (Total) | 50 | 50 | 50 | 50 | 15 | 10 | |
| For 100 DPI | 20 | 20 | 20 | 20 | 5 | ||
| For 170 DPI | 20 | 20 | 20 | 20 | 5 | ||
| For survival | 10 | 10 | 10 | 10 | 5 | 10 | |
Mean hazard ratios with 95% confidence intervals generated from the Kaplan-Meier curves
| Doses | Compounds | DMSO | |
|---|---|---|---|
| BMD42-29 | BMD42-2910 | ||
| 5 mg/kg | 1.39 (0.39~5.11) | 0.59 (0.19~1.89) | 2.67 (0.53~13.61) |
| 20 mg/kg | 1.94 (4.49~7.95) | 0.46 (0.15~1.47) | |
References
- Prusiner SB (1991) Molecular biology of prion diseases. Science 252:1515-1522.


- Prusiner SB (1997) Prion diseases and the BSE crisis. Science 278:245-251.
- Prusiner SB, Scott MR, DeArmond SJ, Cohen FE (1998) Prion protein biology. Cell 93:337-348.
- Bolton DC, McKinley MP, Prusiner SB (1982) Identification of a protein that purifies with the scrapie prion. Science 218:1309-1311.
- McKinley MP, Masiarz FR, Isaacs ST, Hearst JE, Prusiner SB (1983) Resistance of the scrapie agent to inactivation by psoralens. Photochem Photobiol 37:539-545.
- DeArmond SJ, McKinley MP, Barry RA, Braunfeld MB, McColloch JR, Prusiner SB (1985) Identification of prion amyloid filaments in scrapie-infected brain. Cell 41:221-235.
- Basler K, Oesch B, Scott M, Westaway D, Wälchli M, Groth DF, McKinley MP, Prusiner SB, Weissmann C (1986) Scrapie and cellular PrP isoforms are encoded by the same chromosomal gene. Cell 46:417-428.
- Cohen FE, Prusiner SB (1998) Pathologic conformations of prion proteins. Annu Rev Biochem 67:793-819.
- Kocisko DA, Baron GS, Rubenstein R, Chen J, Kuizon S, Caughey B (2003) New inhibitors of scrapie-associated prion protein formation in a library of 2000 drugs and natural products. J Virol 77:10288-10294.

- Cashman NR, Caughey B (2004) Prion diseases--close to effective therapy? Nat Rev Drug Discov 3:874-884.
- Cardinale A, Filesi I, Vetrugno V, Pocchiari M, Sy MS, Biocca S (2005) Trapping prion protein in the endoplasmic reticulum impairs PrPC maturation and prevents PrPSc accumulation. J Biol Chem 280:685-694.
- Giles K, Olson SH, Prusiner SB (2017) Developing therapeutics for PrP prion diseases. Cold Spring Harb Perspect Med 7:a023747.
- Teruya K, Doh-Ura K (2017) Insights from therapeutic studies for PrP prion disease. Cold Spring Harb Perspect Med 7:a024430.
- Rossi G, Salmona M, Forloni G, Bugiani O, Tagliavini F (2003) Therapeutic approaches to prion diseases. Clin Lab Med 23:187-208.
- Trevitt CR, Collinge J (2006) A systematic review of prion therapeutics in experimental models. Brain 129:2241-2265.
- Dormont D (2003) Approaches to prophylaxis and therapy. Br Med Bull 66:281-292.
- Otto M, Cepek L, Ratzka P, Doehlinger S, Boekhoff I, Wiltfang J, Irle E, Pergande G, Ellers-Lenz B, Windl O, Kretzschmar HA, Poser S, Prange H (2004) Efficacy of flupirtine on cognitive function in patients with CJD: a double-blind study. Neurology 62:714-718.
- Nakajima M, Yamada T, Kusuhara T, Furukawa H, Takahashi M, Yamauchi A, Kataoka Y (2004) Results of quinacrine administration to patients with Creutzfeldt-Jakob disease. Dement Geriatr Cogn Disord 17:158-163.
- Furlow TW Jr, Whitley RJ, Wilmes FJ (1982) Repeated suppression of Creutzfeldt-Jakob disease with vidarabine. Lancet 2:564-565.
- Tsuboi Y, Doh-Ura K, Yamada T (2009) Continuous intraventricular infusion of pentosan polysulfate: clinical trial against prion diseases. Neuropathology 29:632-636.
- Benito-León J (2004) Combined quinacrine and chlorpromazine therapy in fatal familial insomnia. Clin Neuropharmacol 27:201-203.
- Arruda WO, Bordignon KC, Milano JB, Ramina R (2004) [Creutzfeldt-Jakob disease, Heidenhain variant: case report with MRI (DWI) findings]. Arq Neuropsiquiatr 62:347-352. Portuguese.
- Haïk S, Marcon G, Mallet A, Tettamanti M, Welaratne A, Giaccone G, Azimi S, Pietrini V, Fabreguettes JR, Imperiale D, Cesaro P, Buffa C, Aucan C, Lucca U, Peckeu L, Suardi S, Tranchant C, Zerr I, Houillier C, Redaelli V, Vespignani H, Campanella A, Sellal F, Krasnianski A, Seilhean D, Heinemann U, Sedel F, Canovi M, Gobbi M, Di Fede G, Laplanche JL, Pocchiari M, Salmona M, Forloni G, Brandel JP, Tagliavini F (2014) Doxycycline in Creutzfeldt-Jakob disease: a phase 2, randomised, double-blind, placebo-controlled trial. Lancet Neurol 13:150-158.
- Panegyres PK, Armari E (2013) Therapies for human prion diseases. Am J Neurodegener Dis 2:176-186.
- Hyeon JW, Choi J, Kim SY, Govindaraj RG, Jam Hwang K, Lee YS, An SS, Lee MK, Joung JY, No KT, Lee J (2015) Discovery of novel anti-prion compounds using in silico and in vitro approaches. Sci Rep 5:14944.
- Wang F, Wang X, Orrú CD, Groveman BR, Surewicz K, Abskharon R, Imamura M, Yokoyama T, Kim YS, Vander Stel KJ, Sinniah K, Priola SA, Surewicz WK, Caughey B, Ma J (2017) Self-propagating, protease-resistant, recombinant prion protein conformers with or without in vivo pathogenicity. PLoS Pathog 13:e1006491.
- Choi J, Govindaraj RG, Hyeon JW, Lee K, Ma S, Kim SY, Lee J, No KT (2017) Structural insight into the antiprion compound inhibition mechanism of native prion folding over misfolding. Chem Biol Drug Des 89:907-917.
- Wilham JM, Orrú CD, Bessen RA, Atarashi R, Sano K, Race B, Meade-White KD, Taubner LM, Timmes A, Caughey B (2010) Rapid end-point quantitation of prion seeding activity with sensitivity comparable to bioassays. PLoS Pathog 6:e1001217.
- Cramm M, Schmitz M, Karch A, Zafar S, Varges D, Mitrova E, Schroeder B, Raeber A, Kuhn F, Zerr I (2015) Characteristic CSF prion seeding efficiency in humans with prion diseases. Mol Neurobiol 51:396-405.
- Hyeon JW, Kim SY, Lee SM, Lee J, An SS, Lee MK, Lee YS (2017) Anti-prion screening for acridine, dextran, and tannic acid using real time-quaking induced conversion: a comparison with PrPSc-infected cell screening. PLoS One 12:e0170266.
- Titlow WB, Lee CH, Ryou C (2013) Characterization of toxicological properties of L-lysine polymers in CD-1 mice. J Microbiol Biotechnol 23:1015-1022.
- Jang CH, Lee S, Park IY, Song A, Moon C, Cho GW (2019) Memantine attenuates salicylate-induced tinnitus possibly by reducing NR2B expression in auditory cortex of rat. Exp Neurobiol 28:495-503.
- Asuni AA, Hilton K, Siskova Z, Lunnon K, Reynolds R, Perry VH, O'Connor V (2010) Alpha-synuclein deficiency in the C57BL/6JOlaHsd strain does not modify disease progression in the ME7-model of prion disease. Neuroscience 165:662674.
- Hwang DH, Park HH, Shin HY, Cui Y, Kim BG (2018) Insulinlike growth factor-1 receptor dictates beneficial effects of treadmill training by regulating survival and migration of neural stem cell grafts in the injured spinal cord. Exp Neurobiol 27:489-507.
- Simon D, Herva ME, Benitez MJ, Garrido JJ, Rojo AI, Cuadrado A, Torres JM, Wandosell F (2014) Dysfunction of the PI3K-Akt-GSK-3 pathway is a common feature in cell culture and in vivo models of prion disease. Neuropathol Appl Neurobiol 40:311-326.
- Leidel F, Eiden M, Geissen M, Hirschberger T, Tavan P, Giese A, Kretzschmar HA, Schätzl H, Groschup MH (2014) Piperazine derivatives inhibit PrP/PrP(res) propagation in vitro and in vivo. Biochem Biophys Res Commun 445:23-29.
- Leidel F, Eiden M, Geissen M, Kretzschmar HA, Giese A, Hirschberger T, Tavan P, Schätzl HM, Groschup MH (2011) Diphenylpyrazole-derived compounds increase survival time of mice after prion infection. Antimicrob Agents Chemother 55:4774-4781.
- Pagadala NS, Perez-Pineiro R, Wishart DS, Tuszynski JA (2015) In silico studies and fluorescence binding assays of potential anti-prion compounds reveal an important binding site for prion inhibition from PrP(C) to PrP(Sc). Eur J Med Chem 91:118-131.
- Hosokawa-Muto J, Kamatari YO, Nakamura HK, Kuwata K (2009) Variety of antiprion compounds discovered through an in silico screen based on cellular-form prion protein structure:correlation between antiprion activity and binding affinity. Antimicrob Agents Chemother 53:765-771.
- Kamatari YO, Hayano Y, Yamaguchi K, Hosokawa-Muto J, Kuwata K (2013) Characterizing antiprion compounds based on their binding properties to prion proteins: implications as medical chaperones. Protein Sci 22:22-34.
- Touil F, Pratt S, Mutter R, Chen B (2006) Screening a library of potential prion therapeutics against cellular prion proteins and insights into their mode of biological activities by surface plasmon resonance. J Pharm Biomed Anal 40:822-832.
- Vilette D (2008) Cell models of prion infection. Vet Res 39:10.
- Béranger F, Mangé A, Solassol J, Lehmann S (2001) Cell culture models of transmissible spongiform encephalopathies. Biochem Biophys Res Commun 289:311-316.
- Kang HE, Mo Y, Abd Rahim R, Lee HM, Ryou C (2017) Prion diagnosis: application of real-time quaking-induced conversion. Biomed Res Int 2017:5413936.
- Turner PV, Brabb T, Pekow C, Vasbinder MA (2011) Administration of substances to laboratory animals: routes of administration and factors to consider. J Am Assoc Lab Anim Sci 50:600-613.
- Rubenstein R, Chang B (2013) Re-assessment of PrP(Sc) distribution in sporadic and variant CJD. PLoS One 8:e66352.
- Kuwata K, Nishida N, Matsumoto T, Kamatari YO, HosokawaMuto J, Kodama K, Nakamura HK, Kimura K, Kawasaki M, Takakura Y, Shirabe S, Takata J, Kataoka Y, Katamine S (2007) Hot spots in prion protein for pathogenic conversion. Proc Natl Acad Sci U S A 104:11921-11926.
- Miller-Vedam L, Ghaemmaghami S (2013) Strain specificity and drug resistance in anti-prion therapy. Curr Top Med Chem 13:2397-2406.