Articles

Article Tools
 View Full Text View Full Text |
 Abstract Abstract |
 Article as PDF Article as PDF |
 Print this Article Print this Article |
 Pubmed Pubmed |
 PMC PMC |
 PubReader PubReader |
 Export to Citation Export to Citation |
 Email Alerts Email Alerts |
 Open Access Open Access |
Share this article on :



Stats or Metrics
Article
Review Article
Exp Neurobiol 2013; 22(3): 133-142
Published online September 30, 2013
https://doi.org/10.5607/en.2013.22.3.133
© The Korean Society for Brain and Neural Sciences
Neuronal Autophagy and Neurodevelopmental Disorders
Kyung-Min Lee1, Su-Kyung Hwang2 and Jin-A Lee3*
1Department of Anatomy, Graduate School of Medicine, Kyungpook National University, 2Department of Pediatrics, Kyungpook National University Hospital, Daegu 700-422, 3Department of Biological Science and Biotechnology, College of Life Science and Nanotechnology, Hannam University, Dajeon 305-811, Korea
Correspondence to: *To whom correspondence should be addressed.
TEL: 82-42-629-8785, FAX: 82-42-629-8769
e-mail: leeja@hnu.kr
Abstract
Neurodevelopmental disorders include a wide range of diseases such as autism spectrum disorders and mental retardation. Mutations in several genes that regulate neural development and synapse function have been identified in neurodevelopmental disorders. Interestingly, some affected genes and pathways in these diseases are associated with the autophagy pathway. Autophagy is a complex, bulky degradative process that involves the sequestration of cellular proteins, RNA, lipids, and cellular organelles into lysosomes. Despite recent progress in elucidating the genetics and molecular pathogenesis of these disorders, little is known about the pathogenic mechanisms and autophagy-related pathways involved in common neurodevelopmental disorders. Therefore, in this review, we focus on the current understanding of neuronal autophagy as well as recent findings on genetics and the roles of autophagy pathway in common neurodevelopmental disorders.
Keywords: mTOR, autophagy, neurodevelopment, homeostasis, neurodevelopmental disorders
INTRODUCTION
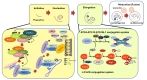
Autophagy, which is a highly conserved pathway from yeast to mammals, is a major catabolic process that delivers cytosolic components to lysosomes for degradation. It is considered to be important for cellular homeostasis, especially under nutrient-deficient or stress conditions, by degrading cytosolic materials in order to either supply the components required for alternate energy metabolism pathways or remove toxic components for cell survival. However, a growing body of evidence has suggested that autophagy is constitutively activated during normal nutrient conditions in a cell-type specific manner. Autophagy has been implicated in various cellular processes such as protein and organelle quality control, development and differentiation, ageing, and immunity. Therefore, alteration of autophagy is associated with several cellular pathologies and diseases, including tumor formation, infectious diseases, liver diseases, myopathy, diabetes, and several neurodegenerative diseases [1, 2]. Autophagy can be generally classified as microautophagy, chaperone-mediated (CMA), or macroautophagy [2-4]. Microautophagy delivers the cytoplasmic contents by invagination of the lysosomal membrane into its lumen. CMA involves the selective sequestration of proteins with a KFERQ-like motif into lysosomes via chaperones Hsc70 and LAMP-2A (
Understanding of the molecular pathway of autophagy has been achieved by identifying several autophagy genes (ATG) from yeast to mammals. Autophagy requires several essential steps for lysosomal degradation: initiation (induction), nucleation (formation of isolation membrane), expansion (elongation of autophagosomes), and maturation of autophagosome. Several ATG are involved in each step of autophagy, and mutations in these genes have been found in several human diseases [2-4].
Autophagy can be induced in an mTOR-dependent or -independent manner by diverse input signals such as nutrients, growth factors, Ca2+, ATP, cAMP, hormone, and protein accumulation under physiological or pathological conditions [4]. Interestingly, in the induction stages of autophagy, many signals converge at the level of the mammalian target of rapamycin complex 1 (mTORC1), which consists of mTOR and several other signaling molecules (DEPTOR [
The Beclin1/PIK3C3 complex (Beclin1 [
The elongation step for efficient expansion of the phagophore requires two ubiquitin-like conjugation systems: the ATG12-ATG5-ATG16L1 (ATG12, ATG7, ATG10, ATG5, and ATG16L1) and LC3-PE (
After completion of autophagosome formation, autophagosomes can fuse with endosomes or lysosomes. Although it is unclear how exactly autophagosomes fuse with lysosomes, their fusion is known to require several proteins such as LAMP2, the Rubicon-UVRAG complex, SNAREs (
Although the autophagy pathway is important for most cells in various tissues, it must be tightly regulated in post-mitotic neurons, which are sensitive to the accumulation of toxic proteins/damaged organelles. Neuronal autophagy is important for proper development and neuronal signaling, which ensures the formation of appropriate neuronal connections and their function. Consequently, alteration of autophagy in neurons causes a cellular traffic jam in highly polarized structures during neural development and neuronal functions. Indeed, autophagy is constitutively active in healthy neurons [12-14]. A previous study using neuron-specific
Neuronal homeostasis in developing neurons might be critical for growth or refinement of their polarized structures such as axons and dendrites. Indeed, during development, proper degradation of proteins or organelles is required for structural plasticity. Recent studies have demonstrated a crucial role for autophagy-related genes in the development and maturation of axons, dendrites, and synapses. Mice with a specific deletion of
Neuronal autophagy might regulate synaptic growth or function in polarized structures such as axons and dendrites. In
Besides synaptic function, previous studies have indicated a role for autophagy in neuronal differentiation. Genetic study based on AMBRA1 haploinsufficiency or pharmacological (wortmanin, 3-methyladenine) treatment showed disrupted neurogenesis in cultured cells derived from the olfactory bulb of mice. Treatment with 3-methyladenine also was shown to impair neurogenesis in chicken otic epitilial cells [31]. Moreover, autophagy impairment disrupts neuroblastoma differentiation into neurons as well as glioma stem/progenitor cell differentiation [32], indicating the role of autophagy in neuronal differentiation and neurogenesis. More interestingly, a recent study showed that Sonic hedgehog (Shh), which is a well known neurodevelopment gene, promotes autophagy in hippocampal neurons, indicating a link between Shh (a neurodevelopmental gene) and autophagy pathways [33].
Acute defects in autophagy during early neurodevelopment or synaptic activity may cause impairment of neural differentiation, axon growth, or neuronal signaling, resulting in synaptic malfunction before severe neurodegeneration. Thus, manipulation of the autophagic pathway may represent an important therapeutic strategy for the promotion of neural differentiation, axonal growth, synaptic growth, or regeneration of neurites in neurodevelopmental disorders.
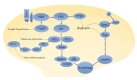
Neurodevelopmental disorders characterized by cognitive deficits and behavioral impairment include a multifaceted group of mental diseases. Numerous works have demonstrated a link between aberrant signaling pathways and various neurodevelopmental disorders represented by abnormal neural structures and function. However, the abnormalities affecting genes and signaling pathways associated with neural development and synaptic function are more collective effects rather than individual ones, as they overlap among various neurodevelopmental disorders. Despite complicated pathophysiological mechanisms underlying neurodevelopmental disorders, the study of autism spectrum disorders (ASDs) has provided insights into the pathways involved in autophagy-related disease pathogenesis. Here, we discuss a subset of ASDs related to mTOR-dependent or -independent signaling associated with autophagy: autism, tuberous sclerosis, and fragile X syndrome and neurofibromatosis (Fig. 2).
Mutations in PTEN (phosphatase and tensin homolog deleted on chromosome ten) are well known to be associated with autism [34]. PTEN functions as a lipid phosphatase to regulate PI3K/Akt signaling and controls TSC-mTOR, which regulates induction of autophagy. Recently, it was reported that disinhibited mTOR is implicated in mice displaying an autistic phenotype, suggesting that autophagy may be deregulated in autism [35]. Parada and colleagues reported that dysfunctional activation of the PI3K/mTOR pathway upon mutation of PTEN is associated with abnormal neuronal arborization and social interaction deficits [35]. Malformation of the neuronal structure resulting from abnormal TSC-mTOR signaling upon PTEN mutation might be associated with alteration of autophagy during neural development. Conversely, pharmacological treatment with rapamycin to inhibit mTOR pathway has been shown to partially restore most autism-like symptoms, including social behaviors, as well as improve abnormal neuroanatomical structures such as neuronal hypertrophy and macrocephaly in PTEN mutant mice [36, 37].
Several autism-associated genes encompass the mTOR-independent Epac2-Rap pathway [38], consistent with a pivotal role in neural development and synaptic plasticity [39]. Epac2, a guanine nucleotide exchange factor (GEF) for Rap (Ras-like small GTPase), is a product of the RAPGEF4 gene responsible for autism [40] and is known to play a role in the inhibition of autophagy. Penzes's research group have reported that Epac2 has an important function in basal dendrite formation during development via Rap signaling, further supporting the involvement of autophagy in neurite growth and Epac2-Rap pathway [38].
Tuberous sclerosis, a type of ASD, is a neurocutaneous syndrome that causes non-malignant tumors in multiple organs. Tuberous sclerosis is caused by mutation of the TSC1 and TSC2 genes, which encode harmartin and tuberin, respectively. A heterodimeric complex consisting of TSC1 and TSC2 regulates protein synthesis in various cells including neurons [41, 42], and retains mTOR in mTORC1 [43-45]. As a protein kinase, mTOR is released from the TSC1/TSC2 complex, which acts as a negative regulator of mTOR, by upstream PI3K and Akt signaling. Patients with tuberous sclerosis generally have heterozygous mutations in either TSC1 or TSC2, as homozygous gene mutations cause embryonic death. Signal transduction via mTOR is known to be required for synaptic plasticity [46], and enhancement of mTOR activity in tuberous sclerosis is involved not only in the impairment of hippocampal mGluR-LTD (long-term depression) [47, 48], but also dendritic spine plasticity, which alters the characteristics of glutamatergic synapses [49, 50]. Mice with heterozygous mutations in TSC1 or TSC2 display cognitive dysfunction without neuroanatomical defects [51]. In contrast, brief treatment of rapamycin in TSC2+/- mice shows reversal of learning deficits along with enhancement of late-phase LTP (long-term potentiation) [52]. Besides these findings, a recent publication by Sabatini and his colleagues reported that the TSC-mTOR signaling pathway is central to the regulation of neural network activity by suppressing inhibitory synapses. Alterations of the excitatory and inhibitory synaptic balance due to deregulation of TSC-mTOR signaling produce the neurological dysfunction with characteristic of tuberous sclerosis [53]. On the other hand, treatment with rapamycin, an mTOR inhibitor, has been shown to cause persistent recovery of pathological neuronal features as well as several behavioral phenotypes, including impaired social interactions in a mouse model of tuberous sclerosis [54, 55].
Fragile X syndrome, the most common known genetic disease among ASDs, is caused by mutation of FMR1, which codes for FMRP (fragile X mental retardation protein). FMRP negatively regulates the translation of numerous mRNAs at synapses and has been suggested to be important for activity-dependent synapse elimination, a key process during postnatal brain development [56-58]. In a mouse model deficient in homologous FMR1 genes, FMRP was shown to mediate the specific impairment of PI3K-Atk signal transduction by upregulating Ras activity, although MEK (extracellular signal-regulated kinase kinase)-ERK (extracellular signal-regulated kinase) signaling downstream of Ras is normal [59]. Additionally, FMR1 knockout (KO) mice show elevated mTOR signaling, leading to dysregulation of the PI3K/Atk/mTOR pathway [60]. Based on the aforementioned role of neuronal autophagy in synaptic growth and function, evidence has suggested a direct correlation between impairment of PI3K-Atk signaling and abnormal dendritic spine morphogenesis as well as selective defects in associative learning in fragile X syndrome [61-66]. A previous study showed that human patients with fragile X syndrome display dysregulation of mTOR signaling, as observed in a FMR1 KO mouse model [67]. Furthermore, from a fundamental pathophysiological point of view, FMR1 KO mice show increased basal protein synthesis [68, 69], which is consistent with the notion of FMRP as a translational repressor, suggesting the possibility of altered neuronal autophagy activity due to disruption of cellular homeostasis or cellular energy metabolism. Indeed, the molecular mechanisms linking functional loss of FMRP1 with excessive synthesis of proteins such as PIKE (PI3K enhancer) through aberrant mGluR-dependent synaptic plasticity are implicated in the enhancement of PI3K/mTOR signaling at synapses [60, 70]. These findings highlight the potential roles of mTOR inhibitors such as rapamycin as therapeutic drugs for treatment of fragile X syndrome [71].
NF1 gene implicated in neurofibromatosis encodes neurofibromin, a GTPase activating protein, which acts as a negative regulator of Ras. Disinhibited Ras-MAPK signaling and mTOR hyperactivity have been observed in the patients with NF1 [72]. Mutations in neurofibromin generate overactivity of Ras, leading to enhanced activation of both PI3K and ERK signaling. The activation of these kinases suppresses the TSC1/TSC2 complex, which releases inhibitory activity of Rheb, leading to mTOR activation. Because of over-activation of Ras signaling caused by NF1 gene mutations, the functional suppression of Ras signaling with genetic or pharmacological approaches attenuates the impairment of synaptic plasticity and learning deficits shown in neurofibromatosis animal model [73, 74]. However, several studies using human and mouse brain analysis suggest that dysregulation of mTOR signaling in neurofibromatosis leads to increased glial cell growth as well as neuroglial progenitor proliferation [75] and gliomagenesis [76]. Interestingly, these are not the common cellular defects shown by mutations of PTEN, TSC1, or TSC2. Indeed, over-proliferation of astrocyte and glioma formation caused by functional inactivation of NF1 gene are associated with hyperactivity of mTOR signaling in a TSC-independent manner [76]. Therefore, it will be interesting to elucidate how each mTOR-related signaling affects disease progression and autophagy pathway.
CONCLUSION & PERSPECTIVES
Insight gained from the above studies has led to the following conclusions: 1) Neuronal autophagy may play important roles in structural refinement of neurite growth or regeneration, neural differentiation, synaptic growth, or synaptic plasticity. 2) Therefore, alteration of autophagy during neurodevelopment and synaptic plasticity might cause abnormal development and synaptic malfunction, leading to neurodevelopmental disorders. 3) Mutation of mTOR-related genes or alteration of mTOR signaling in common neurodevelopmental disorders seems to be associated with dysfunctional autophagy pathway, indicating the potential roles of autophagy in these disorders (Fig. 2). 4) The autophagy-associated pathway, including mTOR signaling or mTOR independent-Rap signaling, might contribute to molecular pathogenesis of common neurodevelopmental disorders. 5) The autophagy pathway might represent a potential valuable therapeutic strategy for treatment of neurodevelopmental disorders.
Although previous studies have highlighted potential roles for autophagy during neurodevelopment and synaptic plasticity, further detailed mechanistic studies are needed. The involvement of autophagy-dependent mTOR signaling in neurodevelopmental disorders should be examined since mTOR pathways are associated with several autophagy-independent pathways. Further, there are many interesting questions that need to be addressed in order to elucidate the regulation of autophagy during neurodevelopment and synaptic function. Such questions include: What are the autophagy substrates in axon terminals, dendrites, and synapses? How is autophagy tightly controlled in highly polarized axons or dendrites in a context-dependent manner? How does autophagy affect neurodevelopmental disorders? Is autophagy detrimental or beneficial to these disorders? How do neurodevelopmental genes affect the autophagy pathway? Further studies aimed at identifying the modifiers or modulators of autophagy regulation in highly polarized neurons will contribute to our understanding of the autophagy pathway during development and synaptic plasticity and will further aid the development of therapeutic strategies for treatment of autophagy-associated neurodevelopmental disorders.
{kind=link}
{kind=link}
References
- Levine B, Mizushima N, Virgin HW. Autophagy in immunity and inflammation. Nature 2011;469:323-335.

- Rubinsztein DC, Codogno P, Levine B. Autophagy modulation as a potential therapeutic target for diverse diseases. Nat Rev Drug Discov 2012;11:709-730.
- Mizushima N, Komatsu M. Autophagy: renovation of cells and tissues. Cell 2011;147:728-741.
- Wirawan E, Vanden Berghe T, Lippens S, Agostinis P, Vandenabeele P. Autophagy: for better or for worse. Cell Res 2012;22:43-61.
- Buchan JR, Kolaitis RM, Taylor JP, Parker R. Eukaryotic stress granules are cleared by autophagy and Cdc48/VCP function. Cell 2013;153:1461-1474.
- Williams A, Sarkar S, Cuddon P, Ttofi EK, Saiki S, Siddiqi FH, Jahreiss L, Fleming A, Pask D, Goldsmith P, O'Kane CJ, Floto RA, Rubinsztein DC. Novel targets for Huntington's disease in an mTOR-independent autophagy pathway. Nat Chem Biol 2008;4:295-305.
- Yang Z, Klionsky DJ. Mammalian autophagy: core molecular machinery and signaling regulation. Curr Opin Cell Biol 2010;22:124-131.
- Klionsky DJ, Bruford EA, Cherry JM, Hodgkin J, Laulederkind SJ, Singer AG. In the beginning there was babble. Autophagy 2012;8:1165-1167.
- Nishida Y, Arakawa S, Fujitani K, Yamaguchi H, Mizuta T, Kanaseki T, Komatsu M, Otsu K, Tsujimoto Y, Shimizu S. Discovery of Atg5/Atg7-independent alternative macroautophagy. Nature 2009;461:654-658.
- Lee JA, Gao FB. Neuronal functions of ESCRTs. Exp Neurobiol 2012;21:9-15.
- Lee JA, Beigneux A, Ahmad ST, Young SG, Gao FB. ESCRT-III dysfunction causes autophagosome accumulation and neurodegeneration. Curr Biol 2007;17:1561-1567.
- Lee JA. Neuronal autophagy: a housekeeper or a fighter in neuronal cell survival?. Exp Neurobiol 2012;21:1-8.
- Nixon RA. Endosome function and dysfunction in Alzheimer's disease and other neurodegenerative diseases. Neurobiol Aging 2005;26:373-382.
- Xilouri M, Stefanis L. Autophagy in the central nervous system: implications for neurodegenerative disorders. CNS Neurol Disord Drug Targets 2010;9:701-719.
- Hara T, Nakamura K, Matsui M, Yamamoto A, Nakahara Y, Suzuki-Migishima R, Yokoyama M, Mishima K, Saito I, Okano H, Mizushima N. Suppression of basal autophagy in neural cells causes neurodegenerative disease in mice. Nature 2006;441:885-889.
- Komatsu M, Waguri S, Chiba T, Murata S, Iwata J, Tanida I, Ueno T, Koike M, Uchiyama Y, Kominami E, Tanaka K. Loss of autophagy in the central nervous system causes neurodegeneration in mice. Nature 2006;441:880-884.
- Mizushima N, Levine B. Autophagy in mammalian development and differentiation. Nat Cell Biol 2010;12:823-830.
- Nixon RA, Cataldo AM. Lysosomal system pathways: genes to neurodegeneration in Alzheimer's disease. J Alzheimers Dis 2006;9:277-289.
- Song JW, Misgeld T, Kang H, Knecht S, Lu J, Cao Y, Cotman SL, Bishop DL, Lichtman JW. Lysosomal activity associated with developmental axon pruning. J Neurosci 2008;28:8993-9001.
- Coupé B, Ishii Y, Dietrich MO, Komatsu M, Horvath TL, Bouret SG. Loss of autophagy in pro-opiomelanocortin neurons perturbs axon growth and causes metabolic dysregulation. Cell Metab 2012;15:247-255.
- Ban BK, Jun MH, Ryu HH, Jang DJ, Ahmad ST, Lee JA. Autophagy negatively regulates early axon growth in cortical neurons. Mol Cell Biol 2013;33:3907-3919.
- Shi SH, Jan LY, Jan YN. Hippocampal neuronal polarity specified by spatially localized mPar3/mPar6 and PI 3-kinase activity. Cell 2003;112:63-75.
- Jiang H, Guo W, Liang X, Rao Y. Both the establishment and the maintenance of neuronal polarity require active mechanisms: critical roles of GSK-3beta and its upstream regulators. Cell 2005;120:123-135.
- Tee AR, Fingar DC, Manning BD, Kwiatkowski DJ, Cantley LC, Blenis J. Tuberous sclerosis complex-1 and -2 gene products function together to inhibit mammalian target of rapamycin (mTOR)-mediated downstream signaling. Proc Natl Acad Sci U S A 2002;99:13571-13576.
- Kwiatkowski DJ, Manning BD. Tuberous sclerosis: a GAP at the crossroads of multiple signaling pathways. Hum Mol Genet 2005;14 Spec No. 2:R251-R258.
- Choi YJ, Di Nardo A, Kramvis I, Meikle L, Kwiatkowski DJ, Sahin M, He X. Tuberous sclerosis complex proteins control axon formation. Genes Dev 2008;22:2485-2495.
- Tomoda T, Bhatt RS, Kuroyanagi H, Shirasawa T, Hatten ME. A mouse serine/threonine kinase homologous to C. elegans UNC51 functions in parallel fiber formation of cerebellar granule neurons. Neuron 1999;24:833-846.
- Shen W, Ganetzky B. Nibbling away at synaptic development. Autophagy 2010;6:168-169.
- Hernandez D, Torres CA, Setlik W, Cebrián C, Mosharov EV, Tang G, Cheng HC, Kholodilov N, Yarygina O, Burke RE, Gershon M, Sulzer D. Regulation of presynaptic neurotransmission by macroautophagy. Neuron 2012;74:277-284.
- Shehata M, Matsumura H, Okubo-Suzuki R, Ohkawa N, Inokuchi K. Neuronal stimulation induces autophagy in hippocampal neurons that is involved in AMPA receptor degradation after chemical long-term depression. J Neurosci 2012;32:10413-10422.
- Aburto MR, Sánchez-Calderón H, Hurlé JM, Varela-Nieto I, Magariños M. Early otic development depends on autophagy for apoptotic cell clearance and neural differentiation. Cell Death Dis 2012;3:e394.
- Zhao Y, Huang Q, Yang J, Lou M, Wang A, Dong J, Qin Z, Zhang T. Autophagy impairment inhibits differentiation of glioma stem/progenitor cells. Brain Res 2010;1313:250-258.
- Petralia RS, Schwartz CM, Wang YX, Kawamoto EM, Mattson MP, Yao PJ. Sonic hedgehog promotes autophagy in hippocampal neurons. Biol Open 2013;2:499-504.
- Butler MG, Dasouki MJ, Zhou XP, Talebizadeh Z, Brown M, Takahashi TN, Miles JH, Wang CH, Stratton R, Pilarski R, Eng C. Subset of individuals with autism spectrum disorders and extreme macrocephaly associated with germline PTEN tumour suppressor gene mutations. J Med Genet 2005;42:318-321.
- Kwon CH, Luikart BW, Powell CM, Zhou J, Matheny SA, Zhang W, Li Y, Baker SJ, Parada LF. Pten regulates neuronal arborization and social interaction in mice. Neuron 2006;50:377-388.
- Zhou J, Blundell J, Ogawa S, Kwon CH, Zhang W, Sinton C, Powell CM, Parada LF. Pharmacological inhibition of mTORC1 suppresses anatomical, cellular, and behavioral abnormalities in neural-specific Pten knock-out mice. J Neurosci 2009;29:1773-1783.
- Kwon CH, Zhu X, Zhang J, Baker SJ. mTor is required for hypertrophy of Pten-deficient neuronal soma in vivo. Proc Natl Acad Sci U S A 2003;100:12923-12928.
- Srivastava DP, Woolfrey KM, Jones KA, Anderson CT, Smith KR, Russell TA, Lee H, Yasvoina MV, Wokosin DL, Ozdinler PH, Shepherd GM, Penzes P. An autism-associated variant of Epac2 reveals a role for Ras/Epac2 signaling in controlling basal dendrite maintenance in mice. PLoS Biol 2012;10:e1001350.
- Zhu JJ, Qin Y, Zhao M, Van Aelst L, Malinow R. Ras and Rap control AMPA receptor trafficking during synaptic plasticity. Cell 2002;110:443-455.
- Bacchelli E, Blasi F, Biondolillo M, Lamb JA, Bonora E, Barnby G, Parr J, Beyer KS, Klauck SM, Poustka A, Bailey AJ, Monaco AP, Maestrini E, International Molecular Genetic Study of Autism Consortium (IMGSAC). Screening of nine candidate genes for autism on chromosome 2q reveals rare nonsynonymous variants in the cAMP-GEFII gene. Mol Psychiatry 2003;8:916-924.
- Han JM, Sahin M. TSC1/TSC2 signaling in the CNS. FEBS Lett 2011;585:973-980.
- Orlova KA, Crino PB. The tuberous sclerosis complex. Ann N Y Acad Sci 2010;1184:87-105.
- Wienecke R, Maize JC, Shoarinejad F, Vass WC, Reed J, Bonifacino JS, Resau JH, de Gunzburg J, Yeung RS, DeClue JE. Co-localization of the TSC2 product tuberin with its target Rap1 in the Golgi apparatus. Oncogene 1996;13:913-923.
- Castro AF, Rebhun JF, Clark GJ, Quilliam LA. Rheb binds tuberous sclerosis complex 2 (TSC2) and promotes S6 kinase activation in a rapamycin- and farnesylation-dependent manner. J Biol Chem 2003;278:32493-32496.
- Yu Y, Li S, Xu X, Li Y, Guan K, Arnold E, Ding J. Structural basis for the unique biological function of small GTPase RHEB. J Biol Chem 2005;280:17093-17100.
- Tang SJ, Reis G, Kang H, Gingras AC, Sonenberg N, Schuman EM. A rapamycin-sensitive signaling pathway contributes to long-term synaptic plasticity in the hippocampus. Proc Natl Acad Sci U S A 2002;99:467-472.
- Bateup HS, Takasaki KT, Saulnier JL, Denefrio CL, Sabatini BL. Loss of Tsc1 in vivo impairs hippocampal mGluR-LTD and increases excitatory synaptic function. J Neurosci 2011;31:8862-8869.
- von der Brelie C, Waltereit R, Zhang L, Beck H, Kirschstein T. Impaired synaptic plasticity in a rat model of tuberous sclerosis. Eur J Neurosci 2006;23:686-692.
- Tavazoie SF, Alvarez VA, Ridenour DA, Kwiatkowski DJ, Sabatini BL. Regulation of neuronal morphology and function by the tumor suppressors Tsc1 and Tsc2. Nat Neurosci 2005;8:1727-1734.
- Nie D, Di Nardo A, Han JM, Baharanyi H, Kramvis I, Huynh T, Dabora S, Codeluppi S, Pandolfi PP, Pasquale EB, Sahin M. Tsc2-Rheb signaling regulates EphA-mediated axon guidance. Nat Neurosci 2010;13:163-172.
- Goorden SM, van Woerden GM, van der Weerd L, Cheadle JP, Elgersma Y. Cognitive deficits in Tsc1+/- mice in the absence of cerebral lesions and seizures. Ann Neurol 2007;62:648-655.
- Ehninger D, Han S, Shilyansky C, Zhou Y, Li W, Kwiatkowski DJ, Ramesh V, Silva AJ. Reversal of learning deficits in a Tsc2+/- mouse model of tuberous sclerosis. Nat Med 2008;14:843-848.
- Bateup HS, Johnson CA, Denefrio CL, Saulnier JL, Kornacker K, Sabatini BL. Excitatory/inhibitory synaptic imbalance leads to hippocampal hyperexcitability in mouse models of tuberous sclerosis. Neuron 2013;78:510-522.
- Meikle L, Pollizzi K, Egnor A, Kramvis I, Lane H, Sahin M, Kwiatkowski DJ. Response of a neuronal model of tuberous sclerosis to mammalian target of rapamycin (mTOR) inhibitors: effects on mTORC1 and Akt signaling lead to improved survival and function. J Neurosci 2008;28:5422-5432.
- Sato A, Kasai S, Kobayashi T, Takamatsu Y, Hino O, Ikeda K, Mizuguchi M. Rapamycin reverses impaired social interaction in mouse models of tuberous sclerosis complex. Nat Commun 2012;3:1292.
- O'Donnell WT, Warren ST. A decade of molecular studies of fragile X syndrome. Annu Rev Neurosci 2002;25:315-338.
- Pfeiffer BE, Zang T, Wilkerson JR, Taniguchi M, Maksimova MA, Smith LN, Cowan CW, Huber KM. Fragile X mental retardation protein is required for synapse elimination by the activity-dependent transcription factor MEF2. Neuron 2010;66:191-197.
- Zalfa F, Giorgi M, Primerano B, Moro A, Di Penta A, Reis S, Oostra B, Bagni C. The fragile X syndrome protein FMRP associates with BC1 RNA and regulates the translation of specific mRNAs at synapses. Cell 2003;112:317-327.
- Hu H, Qin Y, Bochorishvili G, Zhu Y, van Aelst L, Zhu JJ. Ras signaling mechanisms underlying impaired GluR1-dependent plasticity associated with fragile X syndrome. J Neurosci 2008;28:7847-7862.
- Sharma A, Hoeffer CA, Takayasu Y, Miyawaki T, McBride SM, Klann E, Zukin RS. Dysregulation of mTOR signaling in fragile X syndrome. J Neurosci 2010;30:694-702.
- The Dutch-Belgian Fragile X Consortium. Fmr1 knockout mice: a model to study fragile X mental retardation. Cell 1994;78:23-33.
- Frankland PW, Wang Y, Rosner B, Shimizu T, Balleine BW, Dykens EM, Ornitz EM, Silva AJ. Sensorimotor gating abnormalities in young males with fragile X syndrome and Fmr1-knockout mice. Mol Psychiatry 2004;9:417-425.
- Zhao MG, Toyoda H, Ko SW, Ding HK, Wu LJ, Zhuo M. Deficits in trace fear memory and long-term potentiation in a mouse model for fragile X syndrome. J Neurosci 2005;25:7385-7392.
- Comery TA, Harris JB, Willems PJ, Oostra BA, Irwin SA, Weiler IJ, Greenough WT. Abnormal dendritic spines in fragile X knockout mice: maturation and pruning deficits. Proc Natl Acad Sci U S A 1997;94:5401-5404.
- Irwin SA, Patel B, Idupulapati M, Harris JB, Crisostomo RA, Larsen BP, Kooy F, Willems PJ, Cras P, Kozlowski PB, Swain RA, Weiler IJ, Greenough WT. Abnormal dendritic spine characteristics in the temporal and visual cortices of patients with fragile-X syndrome: a quantitative examination. Am J Med Genet 2001;98:161-167.
- Nimchinsky EA, Oberlander AM, Svoboda K. Abnormal development of dendritic spines in FMR1 knock-out mice. J Neurosci 2001;21:5139-5146.
- Hoeffer CA, Sanchez E, Hagerman RJ, Mu Y, Nguyen DV, Wong H, Whelan AM, Zukin RS, Klann E, Tassone F. Altered mTOR signaling and enhanced CYFIP2 expression levels in subjects with fragile X syndrome. Genes Brain Behav 2012;11:332-341.
- Osterweil EK, Krueger DD, Reinhold K, Bear MF. Hypersensitivity to mGluR5 and ERK1/2 leads to excessive protein synthesis in the hippocampus of a mouse model of fragile X syndrome. J Neurosci 2010;30:15616-15627.
- Qin M, Kang J, Burlin TV, Jiang C, Smith CB. Postadolescent changes in regional cerebral protein synthesis: an in vivo study in the FMR1 null mouse. J Neurosci 2005;25:5087-5095.
- Darnell JC, Van Driesche SJ, Zhang C, Hung KY, Mele A, Fraser CE, Stone EF, Chen C, Fak JJ, Chi SW, Licatalosi DD, Richter JD, Darnell RB. FMRP stalls ribosomal translocation on mRNAs linked to synaptic function and autism. Cell 2011;146:247-261.
- Chuang SC, Zhao W, Bauchwitz R, Yan Q, Bianchi R, Wong RK. Prolonged epileptiform discharges induced by altered group I metabotropic glutamate receptor-mediated synaptic responses in hippocampal slices of a fragile X mouse model. J Neurosci 2005;25:8048-8055.
- Klose A, Ahmadian MR, Schuelke M, Scheffzek K, Hoffmeyer S, Gewies A, Schmitz F, Kaufmann D, Peters H, Wittinghofer A, Nürnberg P. Selective disactivation of neurofibromin GAP activity in neurofibromatosis type 1. Hum Mol Genet 1998;7:1261-1268.
- Costa RM, Federov NB, Kogan JH, Murphy GG, Stern J, Ohno M, Kucherlapati R, Jacks T, Silva AJ. Mechanism for the learning deficits in a mouse model of neurofibromatosis type 1. Nature 2002;415:526-530.
- Li W, Cui Y, Kushner SA, Brown RA, Jentsch JD, Frankland PW, Cannon TD, Silva AJ. The HMG-CoA reductase inhibitor lovastatin reverses the learning and attention deficits in a mouse model of neurofibromatosis type 1. Curr Biol 2005;15:1961-1967.
- Lee DY, Yeh TH, Emnett RJ, White CR, Gutmann DH. Neurofibromatosis-1 regulates neuroglial progenitor proliferation and glial differentiation in a brain region-specific manner. Genes Dev 2010;24:2317-2329.
- Banerjee S, Crouse NR, Emnett RJ, Gianino SM, Gutmann DH. Neurofibromatosis-1 regulates mTOR-mediated astrocyte growth and glioma formation in a TSC/Rheb-independent manner. Proc Natl Acad Sci U S A 2011;108:15996-16001.