Articles

Article Tools
 View Full Text View Full Text |
 Abstract Abstract |
 Article as PDF Article as PDF |
 Print this Article Print this Article |
 Pubmed Pubmed |
 PMC PMC |
 PubReader PubReader |
 Export to Citation Export to Citation |
 Email Alerts Email Alerts |
 Open Access Open Access |
Share this article on :



Stats or Metrics
Article
Review Article
Exp Neurobiol 2015; 24(4): 285-300
Published online December 30, 2015
https://doi.org/10.5607/en.2015.24.4.285
© The Korean Society for Brain and Neural Sciences
Exploring the Validity of Valproic Acid Animal Model of Autism
Darine Froy N. Mabunga1, Edson Luck T. Gonzales1, Ji-woon Kim1, Ki Chan Kim1 and Chan Young Shin1,2*
1Department of Neuroscience, School of Medicine, and Neuroscience Research Center, SMART-IABS and KU Open Innovation Center, Konkuk University, 2Department of Pharmacology, School of Medicine, Konkuk University, Seoul 05029, Korea
Correspondence to: *To whom correspondence should be addressed.
TEL: 82-2-2030-7834, FAX: 82-2-2049-7899
e-mail: chanyshin@kku.ac.kr
Abstract
The valproic acid (VPA) animal model of autism spectrum disorder (ASD) is one of the most widely used animal model in the field. Like any other disease models, it can't model the totality of the features seen in autism. Then, is it valid to model autism? This model demonstrates many of the structural and behavioral features that can be observed in individuals with autism. These similarities enable the model to define relevant pathways of developmental dysregulation resulting from environmental manipulation. The uncovering of these complex pathways resulted to the growing pool of potential therapeutic candidates addressing the core symptoms of ASD. Here, we summarize the validity points of VPA that may or may not qualify it as a valid animal model of ASD.
Keywords: autism spectrum disorder, construct validity, face validity, predictive validity, animal model, social communication deficit
INTRODUCTION
Autism spectrum disorder (ASD) is characterized as a pervasive developmental disorder with two core symptoms: social communication deficit and repetitive or restricted behaviors or interests. These core symptoms can be observed before three years of age and individuals with autism usually show various comorbid symptoms such as epilepsy, hyperactivity, attention deficit, mental retardation, sleep problems, gastrointestinal problems, and hypo- or hyper-sensory problems, among others [1,2,3,4,5,6,7,8]. Deciphering the complexities of ASD is becoming one of the biggest challenges for many neuroscientists in the recent years. The number of patients has strikingly increased in the last ten years, and now its prevalence has reached 1 to 1.5% [9]. Despite its increasing prevalence, ASD is still a disorder with a poorly understood pathophysiology and sluggish drug discovery. This can be traced to the complexity of the neurobiology of higher brain functions and the limited accessibility to human brains [10]. Consequentially, there is only one ASD-specific drug approved by the Food and Drug Administration (FDA) to treat the irritability symptoms and, to some extent, repetitive behaviors; but not social communication problems [11,12,13]. Other drugs are prescribed by physicians to manage the accompanying symptoms in ASD patients such as anxiety, depression, inattention, seizures and other behavioral problems [14]. Addressing the heterogeneity of its symptoms may pose as a financial burden to families who are raising an autistic child.
There is substantial progress, however, in the deciphering of ASD's complex pathophysiology and the development of therapeutics. This can be attributed to the critical role played by the animal models. Like any other animal models of neurologic disorders, ASD animal models were developed to gain insight and make predictions of the human ASD condition [15]. Despite progress, the working of these animal models still manages to baffle researchers. The ease of developing these models through genetic modification and environmental exposure has not assuaged the complexity of their mechanisms [10]. Autism is a specific disorder in humans, and most of its symptoms can only be approximated by animal models. This prompts a need to systematically approach the development and evaluation of animal models using a scientific method to validate and consequently improve the confidence in a model.
Chadman [16] and van der Staay [17] summarized the goals of animal models into (a) validating the hypotheses about the mechanisms underlying the disease and (b) evaluating the translational benefit of the therapeutics for the disease. To meet these purposes, an animal model must qualify on several criteria: most commonly, construct, face, and predictive validity [10]. The strength of the model then can be deduced from the commonalities between the animal model and the human disease based on these criteria of validity. The animal model is also further tested with the criteria of reliability, whether quantitative measures agree with the standard statistical analyses, and replicability, whether results can be reproducible in the same laboratory and other laboratories [15]. A number of animal models of ASD exist and are currently being used as a tool in understanding the mechanisms of the disease. Belzung et al [18], in a review, listed four categories of ASD animal models: models of neuropeptide abnormalities, models mimicking epigenetic marks that increase autism risk in humans, models of neonatal lesions on brain areas affected in individuals with autism, and models of human genetic diseases associated with autism. Rodent models of neuropeptide deficit suggest that structurally related neuropeptides oxytocin and vasopressin may play a vital part in processing social cues and social bond formation in both humans and rodents [19]. Substances which cause possible epigenetic modifications such as valproic acid, thalidomide and 5-methoxytryptamine (5-MT) are also used to generate models of autism. Maternal immune activation is also gaining popularity recently, and substances like poly(I:C) are administered to mimic viral infection in pregnant mouse dams [20]. Also, early lesions at different time points in the neonatal period and at different brain areas resulted into animal models showing symptoms reminiscent of ASD.
Within the last decade, the number of researches using the prenatal (or early postnatal) valproic acid (VPA) exposure animal model is increasing. Our laboratory has also given special attention to this model for its strong etiologic relevance [21,22,23,24] and for its versatility. Clinically, VPA is a medication used for epilepsy and mood swings. However, the use of VPA during childbearing years, and particularly during early pregnancy, is of concern. For years, it has been associated with various teratogenic effects and, more recently, increased risk of autism. Reported clinical trials have caused a ripple of pre-clinical studies using VPA to induce autism-like behavior in rats and mice. Recently, Roullet et al. [25] extensively reviewed clinical and animal studies probing
CONSTRUCT VALIDITY
The first on the list of criteria is construct validity. Construct validity relies on the degree of similarity between the mechanisms underlying the animal model and human disease. This requires that the rationale of the behavioral and biological factors underlying the disorder to be similar between animal models and human patients [27,28]. Argued by some to be the most important criterion for animal models [15], construct validity points to the soundness of the established empirical and theoretical relationship of the model to the disease being modeled, rather than the determined causation between a test and an accepted scale [17]. Therefore, the validity provided by a model's construct becomes the framework for interpreting data generated by the other two criteria of validity. Whether one agrees that construct validity is the most important criterion or not, it is critical to be circumspect about the relationships of the manipulations and the measurements to the animal model being tested [29].
Considering human evidence of autism followed by an early teratogenic insult from VPA, Rodier et al. [30] first developed an animal model of autism by exposing rats to VPA
Studies have supposed that oxidative stress could have down-regulated redox-sensitive enzymes involved in a mechanism called the methionine cycle [33,34]. Disruption of this cycle results to decreased synthesis of cysteine and glutathione, thereby disabling normal antioxidant activity [35]. One study [35] observed lower redox ratio of reduced glutathione to oxidized glutathione (consistent with oxidative stress) in children with autism compared to control children. Furthermore, erythrocyte and plasma level of glutathione peroxidase (GSH-Px) activities are lowered in children with autism which have led to the ineffective removal of H2O2 and increased production of highly reactive hydroxyl radicals. ASD patients also have decreased erythrocyte SOD activity that further implies impaired antioxidant defense mechanism [36].
Ornoy [37] has suggested that VPA may create specific embryonic changes and that fetal brains in animals are more sensitive to increments in reactive oxygen species (ROS) than any other fetal organs. This susceptibility to oxidative stress combined with immature embryonic antioxidant defense system may contribute to the resulting teratogenic effect of VPA. Moreover, a study in mice demonstrated that VPA exposure resulted in an increased expression of apoptotic markers [38]. These markers were more visible in the neuroepithelium and were postulated, along with altered embryonic signaling, to be the underlying cause of neural tube defects (NTDs) in the offspring. Also, antioxidants, such as vitamin E and ascorbic acid, which were given as a pretreatment or supplement have been demonstrated to attenuate VPA-induced fetal toxicity and malformations [39,40]. Conformingly, increased oxidative stress was also demonstrated in young children treated with VPA [41]. However, there seems to be no proof yet that embryonic or fetal oxidative stress can be directly induced by VPA.
Another factor gaining attention is the inhibition of histone deacetylase (HDAC), a negative regulator of gene expression, by VPA. A recent study [42] showed that VPA exposure causes a transitory increase in acetylated histone levels in the embryonic brain of mice. This transient hyperacetylation causes an increase in apoptotic cell death in the neocortex and a decrease in cell proliferation in ganglionic imminence. The mice subjected to this study also showed behavioral alterations reminiscent of autism-like behaviors. A study of human embryonic cells demonstrated that VPA inhibits nuclear HDAC activity
Our laboratory's work is currently focused on the downstream signaling pathways involving the HDAC inhibitory action of VPA when exposed prenatally to rats. Firstly, after demonstrating that prenatal VPA exposure causes ASD-like behavior phenotypes in the rat offspring [23], we found that these ASD-inducing effects could be traced back to the acetylation of gene promoter regions in the embryonic brain after VPA exposure. Indeed, VPA injection at embryonic day 12 (E12) induced the increased acetylation of Pax6 promoter region of embryonic brains, which caused early and transient overexpression of paired box 6 (Pax6), a gene implicated in sequential glutamatergic differentiation [44]. As a result, glutamatergic transcription promoters, such as Ngn2, Tbe2, and NeuroD1, were sequentially overexpressed after Pax6 initiation, leading to the increased markers of glutamatergic neurons during development [24]. Further confirming these results are the reduced sequential expression of glutamatergic precursors and reduced neuronal markers as a result of the knockdown of Pax6 through RNA interference using siRNA. Also, to justify that the HDAC inhibition of VPA causes Pax6 overexpression, we demonstrated that valpromide, a VPA analog without HDAC inhibitory property, did not initiate Pax6 overexpression in the embryonic brain [24]. Kataoka et al. [42] also demonstrated similar results where VPA prenatal exposure in mice, and not valpromide, initiated a transient histone hyperacetylation in the embryonic brain.
Overall, the role of the HDAC inhibitory activity of VPA has been elucidated in our studies. Various avenues for pathophysiologic tracing can be inferred, with some genetic and, even, epigenetic changes discovered in the VPA model. These areas are essential for the development of ASD therapeutics. For example, based on the increased glutamatergic differentiation in VPA-exposed animals, one can assume that glutamatergic antagonist may have therapeutic potential in this model. Actually, treatment with NMDA receptor antagonists MK-801 and memantine rescued the social impairments and electric seizure sensitivity in the VPA-exposed offspring [24]. Thus, genetic analysis and imaging studies in humans combined with preclinical results in the VPA animal model would be helpful tools to develop patient-specific therapeutics for ASD.
Another important finding from the VPA animal model is the gender bias of ASD phenotypes to the male offspring [21,22], which also mimics the clinical prevalence. Only male offspring showed social impairment, decreased electroseizure threshold and some hyperactivity behaviors [22]. Interestingly, the brains of VPA-exposed male offspring, but not females, have significantly lower MeCP2 expression compared to control levels [21]. The abrogation of MeCP2 expression has been associated with Rett syndrome, which shows ASD-like behavioral phenotypes. Again, we scrutinized that the HDAC inhibitory action of VPA is to be blamed for the decreased MeCP2 expression by testing VPA and its analogs for direct targeting to this gene. Indeed, all VPA analogs, except valpromide which lack HDAC inhibition, reduced the MeCP2 levels
Since its introduction, the E/I imbalance has been a highly recognized theory in ASD based on clinical and preclinical evidences [45,46,47]. Many genetic and environmental factors target specific or general neural system that partly or wholly involves the dysregulation of synaptic formation and transmission [48,49,50], another potential unifying explanation for the complexity of ASD [51]. Of interest, the VPA animal model substantially supports this theory as observed in our studies and in others [21,24,48]. We have explored various pathways in which VPA can induce impairments in neural development, and all those pathways led to the increased glutamatergic neuronal density of the rat brain. This finding was evidenced by macrocephaly, increased neuronal number, and increased protein markers such as PSD95, α-CaMKII, NMDA and AMPA receptors in postnatal VPA rat brains [21,22,24,52,53]. Fukuchi et al. conducted a microarray analysis in cultured rat cortical neurons after treatment with VPA and found hundreds of genes that were affected, either up- or down-regulated [48]. Of particular interest, developmental genes such as BDNF and GABAARA4, involved in epileptogenesis, were upregulated; whereas genes responsible for GABAergic inhibitory neuronal development were downregulated [48]. The VPA animal model, aside from genetic models, is shedding light on the E/I imbalance observed in autism and offers good ground for therapeutic development.
In a study wherein fresh frozen brain tissue from the anterior cingulate cortex (ACC) of diagnosed autistic patients underwent multiple-concentration binding assay, a significant decrease in the mean density of GABAA receptors in the supragranular and infragranular areas of ACC was observed. The authors suggested that this finding might be a result of the increased GABA innervation and release [54]. The same reduction in GABA receptors are also found in the hippocampus [55], indicating widespread GABA receptor abnormalities in ASD.
Hyperserotonemia in autism, a very appealing hypothesis, might also add to the strength of the construct validity of the VPA model. It is considered by many researchers to be the most commonly observed and well-replicated change in autism [56]. At the early stages of development, high plasma levels of serotonin may enter the brain of a developing fetus as blood brain barrier is not yet completely developed. In fact, synthesis of serotonin is high in normally developing young children followed by a slow decline. However, this appears to be defective in individuals with autism such that serotonin levels remain high in their brains [57]. These high levels of serotonin cause a loss of serotonergic terminals through negative feedback, subsequently disrupting serotonergic functions. In pregnant rats exposed to VPA on E9, serotonergic neurons were observed to have abnormally developed in the dorsal raphe nucleus of the offspring at postnatal day 50 [58]. In the same study, in vitro observations showed VPA's disrupting effect on the maturation of serotonergic neurons from embryonic stem cell-derived neuronal progenitors. In a study using SD rats, platelet-rich plasma serotonergic concentrations were found out to be approximately two to three times higher in the VPA-treated group than in the control group. Aside from hyperserotonemia, the study also demonstrated increased serotonin in the hippocampus and increased dopamine in the frontal cortex [59].
FACE VALIDITY
Face validity insists on the recapitulation of disease endophenotypes to be modeled in animals. Behavioral symptoms, neuropathology, neurophysiological functioning, and neurochemical alterations are examples of endophenotypes or given markers that can be modelled by animals [16]. The similarity of these markers is considered as an initial step in identifying potential animal models of neuropsychiatric disorders [60]. However, it is difficult to precisely reproduce a single behavior modeling the human situation, let alone the entirety of the endophenotypes [10].
Van der Staay [15] contested that less importance should be placed on face validity as a criterion for evaluating animal models, citing various factors that may affect the consequences of animal behavior. Even if an animal model has strong face validity, predictive and construct validity must still be established from the model through scientific process [60]. Most of the time, face validity is challenged, leaving the authors the burden to support its proposed legitimacy.
Autism is diagnosed by two behavioral criteria: (a) persistent social communication and interaction deficits and (b) restricted, repetitive patterns of behaviors, interests, or activities [61]; along with other comorbid traits. Moreover, children with fetal valproate syndrome (FVS) were reported to have demonstrated social deficits and spontaneous stereotypy [31,62,63,64]. Belzung and Lemoine [65] proposed that for any animal model to be labeled as a model of autism, the animal must mirror two mentioned core aspects of the symptomatology, at least. Of interest, when VPA was administered to pregnant rats at E12.5, it induced reduced social interaction [23] and stereotypic-like hyperactivity behavior in the offspring [66].
Impairment in social interaction is already widely recognized as the first defining feature of autism [67,68], and it was a study in 1979 that first characterized the quality of social interaction of children with autism, along with impaired speech [69]. When exposed in a social situation, children with autism showed impaired orienting ability of the social stimuli [70], in which two of the most common features are impaired joint attention/engagement and lack of interactive skills to peers and family [71].
Rodents are social species. They are popular subjects for social tests because of their high levels of social interaction and exploration [72]. Crawley et al. [73] reasoned that a quantitative measure of social interaction is an essential factor in establishing an animal model of autism. From a pool of existing literature to score social interaction in animals, Crawley introduced an assay that measures the tendency of the mouse to engage in social interaction with a stranger animal. In a three-chambered apparatus, the mouse is allowed to explore a novel object (wire cage) and a stranger mouse, inside a wire cage placed separately at both ends of the chamber. The time spent in each chamber and time sniffing the wire cage or the caged animal is measured. This set-up prevents direct physical contact between the tested animal and the stimulus animal to measure solely the former's sociability. Bambini-Junior et al., as well as our laboratory, adapted this assay to measure the social interaction and social preference in rats [22,74]. Results from our studies [22,23,24] showed marked decreases in social interaction of VPA-treated rats, consistent with other findings [42,75].
Schneider et al. [66,75] compared sociability of VPA-treated adult and adolescent rats to unfamiliar animals in an open field arena. They reported a decrease in the number of play behavior in VPA adolescent rats while decreased latency to engage in social behavior and reduced social exploration in VPA adult rats. In mice, some social tests were performed in an open-field arena with novel objects or stranger mice as subjects for exploration [42,76]. Results indicated a reduction of social behavior and preference. Our VPA-exposed mice model also showed reduction in sociability and social preference in the three-chamber social approach assay [77]. Additionally, Roullet et al. [78] performed a modified three-chamber social assay in mice and observed a reduced sociability behavior and a decrease in the total number of nose poke in the preference for social novelty.
On the other hand, measurement of verbal communication ability in rodents is virtually impossible. Since animals don't use organized language the way humans do, designing a tool to evaluate communication between animals is challenging. Interestingly, researchers were able to identify that rodents emit some frequency of ultrasonic vocalizations (USV) when they communicate, and these frequencies could vary depending on age, emotional state and environmental factors [79]. It was discovered that pups emit 40-kHz USV when separated from their dams while adult rats emit a low frequency of 22-kHz when they detect danger or aversion and a high frequency of 50-kHz for positive or non-aversive conditions [79]. Through tools measuring the frequency of vocal emissions, researchers can identify whether a certain pup or adult rat is initiating a social response. Indeed, one of the few studies, which measured USV in mice prenatally exposed to VPA, observed disruptions in USV during infancy and adult periods [76]. In the updated DSM-V criteria for diagnosing ASD, social behavior and communication are already considered a combined category where communication can be under the umbrella of the broader aspect of socialization. Thus, the use of USV measurement tool can supplement the results for social behavior assays in VPA animal models.
Diagnoses agree that all individuals with autism display repetitive behaviors including stereotypy, compulsions, obsessions, and echolalia, among others [80,81]. Though repetitive behaviors are not specific to autism, individuals with autism are found with increased severity of compulsions, self-injury [81], stereotypies [82], and sameness of behavior [83]. The overall severity of autism is positively correlated with the overall severity of these behaviors. Also, the heterogeneity of the phenotypes of autism creates a variation of symptoms in every individual [84]. Lam et al., has described the clusters of repetitive behaviors in autism as being associated with distinct profiles of other symptoms [85]. Despite the significance of these features in the disorder, there is still a general wanting of a systematic study of abnormal repetitive behaviors in ASD.
Schneider et al. [75] observed an increase in repetitive/stereotypic-like activity in male VPA rats, described as repeatedly making and breaking of the same photo beam for the time interval of 1/10th of a second when placed in a 44×44 cm cage. In another study, VPA-treated rats also demonstrated increased re-entry of the same previously explored arm in a Y-maze, indicating repetitive tendencies [86]. Repetitive tendencies were measured in the spontaneous alternation paradigm. The test is divided into two trials. The first trial allowed the rat to enter either arm, without reward, and stay for 5 s. The rat is then returned to the start arm, and subsequent arm choice was recorded for the second trial. VPA-treated mice were also observed to display increased repetitive-stereotyped movements [87] and self-grooming time [76,88] in an open field. Increase in marbles buried was exhibited by VPA-treated mice in the marble-burying test, indicating repetitive motor behavior [88]. Remarkably, prenatal VPA exposure induces varying degrees and types of repetitive behaviors in animal models which validates an important behavior marker in clinical autism.
In ASD, both the severity of the core symptoms and the strength of associated comorbid traits have high degrees of variability. Although comorbidities are prevalently seen in individuals with ASD, studies in animal models are more focused on the actual core symptoms of the disease [89].
Anecdotally, patients with autism are said to be hypersensitive to touch. A study done by Cascio et al. [8] reported normal perception of light touch and sensations of warm and cool, but enhanced sensitivity to vibration and thermal pain in the participating adults with high-functioning autism. Impairments in auditory temporal processing was also seen in children with ASD as evidenced by their higher thresholds on the auditory temporal order judgment task [90]. Interestingly, epilepsy is considered variable in autism. However, current reports revealed 25% of ASD patients have comorbid epilepsy [1] and occurs more commonly in autistic patients with intellectual disability [91].
Constipation and chronic diarrhea are the two most frequently reported gastrointestinal problems in children with ASD, with parents reporting significantly more GI problems in children with familial ASD than in their unaffected children [7]. Accordingly, increased severity of autism symptoms is associated with higher tendency to developing GI problems. Finally, patients with autism may also suffer from sleeping problems, like prolonged sleep latency [4,5] and decreased sleep efficiency [5], even night walking [6].
In the VPA model of autism, animal subjects demonstrated increased tactile sensitivity, decreased sensitivity to pain, and diminished acoustic prepulse inhibition [66]. It also displayed a reduced threshold for electroshock [23] and pentylenetetrazole-induced seizures [92]. Kim et al. [93] also reported decreased thickness of tunica mucosa and tunica muscularis in the stomach and ileum of VPA-treated rats. Finally, VPA-treated rats display sleep abnormalities observed as frequent arousal during the light/sleep phase [94]. Similar to the variability of associated symptoms in autistic patients, the presence of other behavioral, emotional, cognitive or physical symptoms in the VPA animal model affirms its face validity.
PREDICTIVE VALIDITY
Lastly, predictive validity, perhaps the most debated criterion, completes the list. Predictive validity, in general, indicates the extrapolation of human behavior through laboratory-manipulated animal behavior [28]. This validity can be divided into two concepts. First, according to some authors, predictive validity requires an animal model to accurately show the same response that human demonstrates when exposed to a certain class of drugs [16]. Second, in drug development, predictive validity also refers to the animal model's ability to identify drugs that may be beneficial to humans [95,96].
It is difficult to validate an animal model of autism using the first concept because of the absence of etiology-directed treatments in the market. For the past decades, the progress of drug discovery for autism has been dragging and is often a result of the slipstream of the psychopharmacology of another psychiatric disorder. Although newer medications are relatively successful in controlling the behavioral aberrations observed in autism (Table 2), attempts to address its core symptoms are just still attempts [reviewed in 97]. The second concept, however, allows researchers to accelerate the progress of drug development through discovering drug targets and testing candidate compounds for efficacy, safety, and pharmacokinetic properties [72].
Evidence of environmental enrichment enhancing the performance of rats in various learning tasks [98,99] and social behavior [100] has led to the hypothesis that it can also be beneficial to brain-damaged animals [101]. Studies resulted in an overwhelming affirmation of this hypothesis [102,103,104]. In humans, exercise and behavioral stimulation are found to enhance brain plasticity and optimize health and cognitive function [reviewed in 105]. Studies involving environmental enrichment, therefore, might be beneficial to neurodevelopmental disorders.
Schneider et al. [101] performed pre- and post-weaning environmental enrichment in VPA rats, as it was shown to induce a strong stimulatory effect on rat development [106,107]. From day 7 to 21, rats underwent intensive multisensory stimulation. After the rats had been weaned, from day 22 to 35, they were housed in a cohort of 12 in a box filled with toys that was changed every two days. Results demonstrated that environmental enrichment alleviated the increased locomotor and stereotypic/repetitive-like activity in VPA-exposed rats, as well as enhanced their social behaviors.
Acetylcholine is associated with a number of neurological processes like cognition, memory, neurotransmitter release, to name a few [108,109,110]. The dysregulation of acetylcholine (ACh), a dynamic mediator of neurotransmission and neuronal development in the brain [108,111], is commonly observed in the brain of many ASD patients [112,113] and is currently being linked to the development of behavioral symptoms in autism. This implication prompted us to further investigate the possible role of ACh in ASD using Donepezil, a cholinergic modulator, which is most useful in dementia associated with Alzheimer's disease. This modulation of the cholinergic system can be a new therapeutic target for ASD.
Using mice models, we administered donepezil hydrochloride monohydrate (0.3 mg/kg bw) intraperitoneally to VPA-treated mice from postnatal day 14 to 40, once daily. We observed no abnormal symptoms or toxic effects in the treated group. Subchronic treatment with donepezil increased the sociability index of mice subjects, measured through an adapted three-chamber assay. Donepezil-treated VPA mice also showed lowered repetitive and hyperactive behavior [77].
The role of histaminergic system in neuropsychopharmacology was first evaluated through testing famotidine, a histamine H2-receptor antagonist in patients with schizophrenia. The result was indicative of improved sociability [114]. This initiated the recent growing interest in the role of histamine as a neurotransmitter and its influence on behavior in physiological and pathological conditions [115,116]. The histaminergic system has become an intriguing pharmacological target leading to the development of drugs that could act on various histamine receptors [117]. Because of the shared symptoms of schizophrenia and autism, studies on the use of histamine receptor modulators in treating autistic behaviors were conducted [118,119], yielding positive results.
Baronio et al. [120] administered H3R antagonist ciproxifan (3 mg/kg) to Swiss mice 30 minutes pre-experiment. In the three-chamber social test, treated mice had increased sociability index; while in the marble burying test, they had reduced repetitive behavior as manifested by the less number of marbles buried. By the time the ciproxifan was administered, most of the CNS developmental changes caused by VPA have reached a functional equilibrium. The authors showed that even at a later stage some of the behavioral aberrations in ASD could still be diminished [120]. In terms of predictive validity, this agent and its target has a potential for developing a new drug for ASD using the VPA animal model although studies regarding the role of histamine in autism are still underway.
Bambini-Junior et al. [121] considered resveratrol, a naturally-occurring polyphenolic compound, as a potential therapeutics for autism because of its purported protective and therapeutic roles in several illnesses [reviewed in 122]. Furthermore, resveratrol is also widely known for its antioxidant and neuroprotective properties [123,124,125].
In their study, pregnant Wistar rats received daily subcutaneous injections of resveratrol (3.6 mg/kg) from E6.5 to E18.5 while VPA was administered in E12.5. Rats prenatally exposed to VPA and resveratrol scored similar to that of the control group in sociability and social novelty preference indices, compared to rats treated only with VPA. Their finding was suggestive of the counteracting effects of resveratrol to the development of autistic-like behaviors induced by VPA. The authors further suggested that a global approach should be taken in investigating the physiological effects of both VPA and resveratrol, as prevention of behavior aberrations of ASD may not only involve a specific brain region but is a result of an all-inclusive process. It is then essential to know whether postnatal resveratrol treatment can rescue the autistic phenotypes of VPA-exposed animals. Moreover, as a natural compound with yet unknown specific target, it might be difficult to assess the reliability and potency of resveratrol as a therapeutic compound, unless target identification studies will be conducted.
Along with the core symptoms of autism, increase in locomotor activity is also observed in VPA-induced animal model of autism. In ADHD, this hyperactive phenotype is a consequence of a dysfunction in the catecholaminergic neuronal system, particularly the dopamine and norepinephrine system [126]. Based on this, we evaluated the effects of methylphenidate and atomoxetine, both first-line drug treatments for ADHD, on the hyperactive phenotype of VPA-induced animal model of autism [127]. However, it was only atomoxetine which lowered the increase in locomotor activity in VPA-exposed rat offspring, and not methylphenidate. Deducing from the difference between methylphenidate and atomoxetine, we suggested that the neurological substrates regulating the hyperactive phenotype of VPA-induced model of ASD are different from that of legitimate ADHD cases. Additional studies are needed to confirm the target of atomoxetine in the brain, like catecholamines, to know how it normalized the locomotor activity in the ASD model.
Due to concerns about the potential teratogenicity of ginseng, only a few studies have been done using its extracts during pregnancy. However, a recent study claimed that ginseng doesn't result to embryo-lethality or teratogenicity in experimental animals [128]. This study and the reports of ginseng extracts attenuating dioxininduced testicular toxicity [129,130] prompted us to investigate the effects of KRG on VPA-induced neurodevelopmental toxicity [131]. Korean red ginseng extracts were orally administered at doses 20, 50, 100, and 200 mg/kg from E10 to E15. There were no deaths and abnormalities found in offspring of dams administered with Korean red ginseng extracts. Moreover, the group administered with 200 mg/kg of the extracts showed the greatest significant improvement of social impairment, as compared to the VPA-exposed rats. We also followed up this study by treating postnatal juvenile VPA-exposed mice with KRG (on submission) to determine whether it can have a therapeutic effect, aside from its protective effect. KRG at 50, 100 or 200 mg/kg were given orally once a day for seven days prior to the battery of experiments and continued until the termination of the experiments. Remarkably, the results have shown 200 mg/kg of KRG as an optimum dose by having rescued the impairments in social and locomotor activities and repetitive behaviors in VPA-exposed mice. It is then important to identify the pathway where KRG normalizes the behavior of mice as a proof of concept for drug development.
CONCLUSION
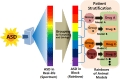
To our knowledge, this is the first review article that discusses in detail the validity of VPA animal model of autism. ASD is extremely heterogeneous in its etiology and presentation of symptoms, which gives it the umbrella term 'Spectrum Disorders'. While it is impossible for the VPA animal model of autism to recapitulate the entire set of signs and symptoms found in individuals with ASD, we still recognize it as an invaluable tool for deciphering the highly-intricate functioning of the human brain and in rushing the discovery of valid treatments, at least for some fraction of the patients sharing the molecular and physiological changes with the model (Fig. 2).
Since autism is a multifactorial disease, all animal models, including the VPA animal model, can only be treated as mimicking an aspect of the entirety of the etiology of autism, termed as a reductionist's view [65]. Truly, not all cases of ASD can be traced to VPA exposure; however, this supposed exposure may share the same pathway with other etiological factors leading to the development of the disease. The model can further take advantage of the current advanced technologies and techniques to confidently outline the mechanistic pathways of VPA's effect to the fetal brain. Making the story more complex, VPA also affects ERK activity, GSK-3beta phosphorylation status, folate cycling, and PPAR-alpha and delta activity, which may in part contribute to the manifestation of autistic traits. The multiplicity of the targets modulated by VPA implicates that care should be taken in data interpretation, especially when investigating the therapeutic potential of candidate molecules.
The VPA model aims to uncover the pathophysiology of the complex ASD from the environmental risk factor perspective, which could also co-occur with the genetic susceptibilities. Additionally, the interaction of both environmental and genetic risk factors for ASD will be an important area of investigation in which the complementary contribution of the two factors could enhance the etiologic strength to achieve a pathologic state in the brain and behavior.
The VPA model offers high face validity since it appropriately replicates, though in approximation, the disturbed behavior in individuals with ASD. Findings in the social behavior paradigms used in VPA animal models were reminiscent of the social deficiency in humans. Stereotypic/repetitive behaviors could also be observed and quantified in both humans and animals with and without stimulation. However, assessment of the increased repetitive behaviors of the VPA animal model varies between laboratories. It is only essential to have a unifying measurement for repetitive behaviors in an animal model to create a standard of evaluation for this symptom. Some comorbidities seen along with the core symptoms of ASD are replicated by the model. The presence of these comorbidities hints that VPA may have global neurologic effects beyond the established scope of ASD. Like any other animal models, the ASD animal model has restrictions in its translation to clinical settings. For example, the extent of the similarities shared between motor stereotypies observed in animals and the more characteristic autistic stereotypies is still an unresolved issue [132]. Also, observing social communication deficits is yet to be expanded, like characterizing the types and social functions of USVs in rodents [133]. Of course, in every VPA animal model, the replication of the core symptoms of ASD precedes the molecular investigations of how VPA induces ASD-like phenotypes. Experimental designs must also be chosen appropriately to effectively link the resemblance of the animal behavior to the human disorder.
To date, pharmacologic treatment addressing the core symptoms of ASD is still elusive, making judgments on predictive validity even more challenging. However, this model proves to be an excellent tool for testing new drug targets and developing novel behavior and drug therapies. Indeed, the results have been encouraging. So far, the possible targets to modulate the social and repetitive behaviors of VPA animal models include NMDA receptor agonists, AChE inhibitors, and histamine antagonist. Interestingly, the preclinical testing of nutraceuticals such as resveratrol and Korean red ginseng, has also yielded positive results in preventing and normalizing the effects VPA in the developing offspring. We can infer that the complementary effects and various targets of the known and yet unknown functions of nutraceuticals would be beneficial to a complex disease such as ASD. Additionally, the testing of other drug entities for the comorbid symptoms of ASD will greatly improve the quality of life of patients, as well as easing the burden of their families.
Overall, as a valid investigative tool, it is only appropriate to focus more on the interpretation of the data extracted from the VPA model of autism, and not on its supposed "truth." These data can be interpreted alongside with the information generated from other models of autism. As van der Staay says, "No animal model can be valid in all situations, in all purposes."
Figures
{kind=link}
{kind=link}
Tables
{kind=link}
Pathways affected by VPA and their supposed mechanism include oxidative stress, histone deacetylase inhibition, excitatory/inhibitory imbalance, and hyperserotonemia.
{kind=link}
Pharmacological treatments, such as donepezil, ciproxifan, resveratrol, atomotexetine and Koreand red ginseng, to VPA animal model of ASD and their effects on animals.
References
- Parmeggiani A, Barcia G, Posar A, Raimondi E, Santucci M, Scaduto MC. Epilepsy and EEG paroxysmal abnormalities in autism spectrum disorders. Brain Dev 2010;32:783-789.

- Sturm H, Fernell E, Gillberg C. Autism spectrum disorders in children with normal intellectual levels: associated impairments and subgroups. Dev Med Child Neurol 2004;46:444-447.
- Williams RS, Hauser SL, Purpura DP, DeLong GR, Swisher CN. Autism and mental retardation: neuropathologic studies performed in four retarded persons with autistic behavior. Arch Neurol 1980;37:749-753.
- Allik H, Larsson JO, Smedje H. Sleep patterns of school-age children with Asperger syndrome or high-functioning autism. J Autism Dev Disord 2006;36:585-595.
- Malow BA, Marzec ML, McGrew SG, Wang L, Henderson LM, Stone WL. Characterizing sleep in children with autism spectrum disorders: a multidimensional approach. Sleep 2006;29:1563-1571.
- Krakowiak P, Goodlin-Jones B, Hertz-Picciotto I, Croen LA, Hansen RL. Sleep problems in children with autism spectrum disorders, developmental delays, and typical development: a population-based study. J Sleep Res 2008;17:197-206.
- Wang LW, Tancredi DJ, Thomas DW. The prevalence of gastrointestinal problems in children across the United States with autism spectrum disorders from families with multiple affected members. J Dev Behav Pediatr 2011;32:351-360.
- Cascio C, McGlone F, Folger S, Tannan V, Baranek G, Pelphrey KA, Essick G. Tactile perception in adults with autism: a multidimensional psychophysical study. J Autism Dev Disord 2008;38:127-137.
- Array. Prevalence of autism spectrum disorder among children aged 8 years-autism and developmental disabilities monitoring network, 11 sites, United States, 2010. MMWR Surveill Summ 2014;63:1-21.
- Nestler EJ, Hyman SE. Animal models of neuropsychiatric disorders. Nat Neurosci 2010;13:1161-1169.
- McDougle CJ, Scahill L, Aman MG, McCracken JT, Tierney E, Davies M, Arnold LE, Posey DJ, Martin A, Ghuman JK, Shah B, Chuang SZ, Swiezy NB, Gonzalez NM, Hollway J, Koenig K, McGough JJ, Ritz L, Vitiello B. Risperidone for the core symptom domains of autism: results from the study by the autism network of the research units on pediatric psychopharmacology. Am J Psychiatry 2005;162:1142-1148.
- McCracken JT, McGough J, Shah B, Cronin P, Hong D, Aman MG, Arnold LE, Lindsay R, Nash P, Hollway J, McDougle CJ, Posey D, Swiezy N, Kohn A, Scahill L, Martin A, Koenig K, Volkmar F, Carroll D, Lancor A, Tierney E, Ghuman J, Gonzalez NM, Grados M, Vitiello B, Ritz L, Davies M, Robinson J, McMahon D, Research Units on Pediatric Psychopharmacology Autism Network. Risperidone in children with autism and serious behavioral problems. N Engl J Med 2002;347:314-321.
- Scahill L, Koenig K, Carroll DH, Pachler M. Risperidone approved for the treatment of serious behavioral problems in children with autism. J Child Adolesc Psychiatr Nurs 2007;20:188-190.
- National Institute of Mental Health. Medications for Autism. Psych Central [Internet]. Bethesda, MD: National Institute of Mental Health; [cited 2015 Sep 21]. ,
Available from: http://psychcentral.com/lib/medications-for-autism/ - van der Staay FJ, Arndt SS, Nordquist RE. Evaluation of animal models of neurobehavioral disorders. Behav Brain Funct 2009;5:11.
- Chadman KK, Yang M, Crawley JN. Criteria for validating mouse models of psychiatric diseases. Am J Med Genet B Neuropsychiatr Genet 2009;150B:1-11.
- van der Staay FJ. Animal models of behavioral dysfunctions: basic concepts and classifications, and an evaluation strategy. Brain Res Brain Res Rev 2006;52:131-159.
- Belzung C, Leman S, Vourc'h P, Andres C. Rodent models for autism: a critical review. Drug Discov Today Dis Models 2005;2:93-101.
- Lim MM, Bielsky IF, Young LJ. Neuropeptides and the social brain: potential rodent models of autism. Int J Dev Neurosci 2005;23:235-243.
- Patterson PH. Immune involvement in schizophrenia and autism: etiology, pathology and animal models. Behav Brain Res 2009;204:313-321.
- Kim KC, Choi CS, Kim JW, Han SH, Cheong JH, Ryu JH, Shin CY. MeCP2 modulates sex differences in the postsynaptic development of the valproate animal model of autism. Mol Neurobiol 2014
- Kim KC, Kim P, Go HS, Choi CS, Park JH, Kim HJ, Jeon SJ, Dela Pena IC, Han SH, Cheong JH, Ryu JH, Shin CY. Male-specific alteration in excitatory post-synaptic development and social interaction in pre-natal valproic acid exposure model of autism spectrum disorder. J Neurochem 2013;124:832-843.
- Kim KC, Kim P, Go HS, Choi CS, Yang SI, Cheong JH, Shin CY, Ko KH. The critical period of valproate exposure to induce autistic symptoms in Sprague-Dawley rats. Toxicol Lett 2011;201:137-142.
- Kim KC, Lee DK, Go HS, Kim P, Choi CS, Kim JW, Jeon SJ, Song MR, Shin CY. Pax6-dependent cortical glutamatergic neuronal differentiation regulates autism-like behavior in prenatally valproic acid-exposed rat offspring. Mol Neurobiol 2014;49:512-528.
- Roullet FI, Lai JK, Foster JA. In utero exposure to valproic acid and autism--a current review of clinical and animal studies. Neurotoxicol Teratol 2013;36:47-56.
- Chomiak T, Turner N, Hu B. What we have learned about autism spectrum disorder from valproic acid. Patholog Res Int 2013;2013:712758.
- Bourin M, Petit-Demoulière B, Dhonnchadha BN, Hascöet M. Animal models of anxiety in mice. Fundam Clin Pharmacol 2007;21:567-574.
- Epstein DH, Preston KL, Stewart J, Shaham Y. Toward a model of drug relapse: an assessment of the validity of the reinstatement procedure. Psychopharmacology (Berl) 2006;189:1-16.
- Lubow RE. Construct validity of the animal latent inhibition model of selective attention deficits in schizophrenia. Schizophr Bull 2005;31:139-153.
- Rodier PM, Ingram JL, Tisdale B, Nelson S, Romano J. Embryological origin for autism: developmental anomalies of the cranial nerve motor nuclei. J Comp Neurol 1996;370:247-261.
- Williams G, King J, Cunningham M, Stephan M, Kerr B, Hersh JH. Fetal valproate syndrome and autism: additional evidence of an association. Dev Med Child Neurol 2001;43:202-206.
- Bescoby-Chambers N, Forster P, Bates G. 'Foetal valproate syndrome and autism: additional evidence of an association'. Dev Med Child Neurol 2001;43:847.
- Gulati S, Chen Z, Brody LC, Rosenblatt DS, Banerjee R. Defects in auxiliary redox proteins lead to functional methionine synthase deficiency. J Biol Chem 1997;272:19171-19175.
- Avila MA, Carretero MV, Rodriguez EN, Mato JM. Regulation by hypoxia of methionine adenosyltransferase activity and gene expression in rat hepatocytes. Gastroenterology 1998;114:364-371.
- James SJ, Cutler P, Melnyk S, Jernigan S, Janak L, Gaylor DW, Neubrander JA. Metabolic biomarkers of increased oxidative stress and impaired methylation capacity in children with autism. Am J Clin Nutr 2004;80:1611-1617.
- Yorbik O, Sayal A, Akay C, Akbiyik DI, Sohmen T. Investigation of antioxidant enzymes in children with autistic disorder. Prostaglandins Leukot Essent Fatty Acids 2002;67:341-343.
- Ornoy A. Valproic acid in pregnancy: how much are we endangering the embryo and fetus?. Reprod Toxicol 2009;28:1-10.
- Tung EW, Winn LM. Valproic acid increases formation of reactive oxygen species and induces apoptosis in postimplantation embryos: a role for oxidative stress in valproic acid-induced neural tube defects. Mol Pharmacol 2011;80:979-987.
- Al Deeb S, Al Moutaery K, Arshaduddin M, Tariq M. Vitamin E decreases valproic acid induced neural tube defects in mice. Neurosci Lett 2000;292:179-182.
- Zhang B, Wang X, Nazarali AJ. Ascorbic acid reverses valproic acid-induced inhibition of hoxa2 and maintains glutathione homeostasis in mouse embryos in culture. Cell Mol Neurobiol 2010;30:137-148.
- Verrotti A, Scardapane A, Franzoni E, Manco R, Chiarelli F. Increased oxidative stress in epileptic children treated with valproic acid. Epilepsy Res 2008;78:171-177.
- Kataoka S, Takuma K, Hara Y, Maeda Y, Ago Y, Matsuda T. Autism-like behaviours with transient histone hyperacetylation in mice treated prenatally with valproic acid. Int J Neuropsychopharmacol 2013;16:91-103.
- Phiel CJ, Zhang F, Huang EY, Guenther MG, Lazar MA, Klein PS. Histone deacetylase is a direct target of valproic acid, a potent anticonvulsant, mood stabilizer, and teratogen. J Biol Chem 2001;276:36734-36741.
- Hevner RF, Hodge RD, Daza RA, Englund C. Transcription factors in glutamatergic neurogenesis: conserved programs in neocortex, cerebellum, and adult hippocampus. Neurosci Res 2006;55:223-233.
- Orekhova EV, Stroganova TA, Prokofyev AO, Nygren G, Gillberg C, Elam M. Sensory gating in young children with autism: relation to age, IQ, and EEG gamma oscillations. Neurosci Lett 2008;434:218-223.
- Casanova MF, Buxhoeveden D, Gomez J. Disruption in the inhibitory architecture of the cell minicolumn: implications for autism. Neuroscientist 2003;9:496-507.
- Rubenstein JL, Merzenich MM. Model of autism: increased ratio of excitation/inhibition in key neural systems. Genes Brain Behav 2003;2:255-267.
- Fukuchi M, Nii T, Ishimaru N, Minamino A, Hara D, Takasaki I, Tabuchi A, Tsuda M. Valproic acid induces up- or down-regulation of gene expression responsible for the neuronal excitation and inhibition in rat cortical neurons through its epigenetic actions. Neurosci Res 2009;65:35-43.
- Bourgeron T. A synaptic trek to autism. Curr Opin Neurobiol 2009;19:231-234.
- Gogolla N, Leblanc JJ, Quast KB, Südhof TC, Fagiolini M, Hensch TK. Common circuit defect of excitatory-inhibitory balance in mouse models of autism. J Neurodev Disord 2009;1:172-181.
- Gogolla N, Leblanc JJ, Quast KB, Südhof TC, Fagiolini M, Hensch TK. Common circuit defect of excitatory-inhibitory balance in mouse models of autism. J Neurodev Disord 2009;1:172-181.
- Go HS, Kim KC, Choi CS, Jeon SJ, Kwon KJ, Han SH, Lee J, Cheong JH, Ryu JH, Kim CH, Ko KH, Shin CY. Prenatal exposure to valproic acid increases the neural progenitor cell pool and induces macrocephaly in rat brain via a mechanism involving the GSK-3β/β-catenin pathway. Neuropharmacology 2012;63:1028-1041.
- Go HS, Seo JE, Kim KC, Han SM, Kim P, Kang YS, Han SH, Shin CY, Ko KH. Valproic acid inhibits neural progenitor cell death by activation of NF-κB signaling pathway and up-regulation of Bcl-XL. J Biomed Sci 2011;18:48.
- Oblak A, Gibbs TT, Blatt GJ. Decreased GABAA receptors and benzodiazepine binding sites in the anterior cingulate cortex in autism. Autism Res 2009;2:205-219.
- Blatt GJ, Fitzgerald CM, Guptill JT, Booker AB, Kemper TL, Bauman ML. Density and distribution of hippocampal neurotransmitter receptors in autism: an autoradiographic study. J Autism Dev Disord 2001;31:537-543.
- McNamara IM, Borella AW, Bialowas LA, Whitaker-Azmitia PM. Further studies in the developmental hyperserotonemia model (DHS) of autism: social, behavioral and peptide changes. Brain Res 2008;1189:203-214.
- Chugani DC. Role of altered brain serotonin mechanisms in autism. Mol Psychiatry 2002;7:S16-S17.
- Miyazaki K, Narita N, Narita M. Maternal administration of thalidomide or valproic acid causes abnormal serotonergic neurons in the offspring: implication for pathogenesis of autism. Int J Dev Neurosci 2005;23:287-297.
- Narita N, Kato M, Tazoe M, Miyazaki K, Narita M, Okado N. Increased monoamine concentration in the brain and blood of fetal thalidomide- and valproic acid-exposed rat: putative animal models for autism. Pediatr Res 2002;52:576-579.
- Geyer MA, Moghaddam B. Animal models relevant to schizophrenia disorders. Neuropsychopharmacology 2002;27:689-701.
- American Psychiatric Association. Diagnostic and statistical manual of mental disorders. 5th ed. Arlington, VA: American Psychiatric Association, 2013.
- Ardinger HH, Atkin JF, Blackston RD, Elsas LJ, Clarren SK, Livingstone S, Flannery DB, Pellock JM, Harrod MJ, Lammer EJ, Majewski F, Schinzel A, Toriello HV, Hanson JW, Optiz JM, Reynolds JF. Verification of the fetal valproate syndrome phenotype. Am J Med Genet 1988;29:171-185.
- Christianson AL, Chesler N, Kromberg JG. Fetal valproate syndrome: clinical and neuro-developmental features in two sibling pairs. Dev Med Child Neurol 1994;36:361-369.
- Laegreid L, Kyllerman M, Hedner T, Hagberg B, Viggedahl G. Benzodiazepine amplification of valproate teratogenic effects in children of mothers with absence epilepsy. Neuropediatrics 1993;24:88-92.
- Belzung C, Lemoine M. Criteria of validity for animal models of psychiatric disorders: focus on anxiety disorders and depression. Biol Mood Anxiety Disord 2011;1:9.
- Schneider T, Przewłocki R. Behavioral alterations in rats prenatally exposed to valproic acid: animal model of autism. Neuropsychopharmacology 2005;30:80-89.
- Volkmar FR, Pauls D. Autism. Lancet 2003;362:1133-1141.
- Lord C, Risi S, Lambrecht L, Cook EH, Leventhal BL, DiLavore PC, Pickles A, Rutter M. The autism diagnostic observation schedule-generic: a standard measure of social and communication deficits associated with the spectrum of autism. J Autism Dev Disord 2000;30:205-223.
- Wing L, Gould J. Severe impairments of social interaction and associated abnormalities in children: epidemiology and classification. J Autism Dev Disord 1979;9:11-29.
- Dawson G, Meltzoff AN, Osterling J, Rinaldi J, Brown E. Children with autism fail to orient to naturally occurring social stimuli. J Autism Dev Disord 1998;28:479-485.
- Kasari C, Patterson S. Interventions addressing social impairment in autism. Curr Psychiatry Rep 2012;14:713-725.
- Kas MJ, Glennon JC, Buitelaar J, Ey E, Biemans B, Crawley J, Ring RH, Lajonchere C, Esclassan F, Talpos J, Noldus LP, Burbach JP, Steckler T. Assessing behavioural and cognitive domains of autism spectrum disorders in rodents: current status and future perspectives. Psychopharmacology (Berl) 2014;231:1125-1146.
- Crawley JN. Mouse behavioral assays relevant to the symptoms of autism. Brain Pathol 2007;17:448-459.
- Bambini-Junior V, Rodrigues L, Behr GA, Moreira JC, Riesgo R, Gottfried C. Animal model of autism induced by prenatal exposure to valproate: behavioral changes and liver parameters. Brain Res 2011;1408:8-16.
- Schneider T, Roman A, Basta-Kaim A, Kubera M, Budziszewska B, Schneider K, Przewłocki R. Gender-specific behavioral and immunological alterations in an animal model of autism induced by prenatal exposure to valproic acid. Psychoneuroendocrinology 2008;33:728-740.
- Gandal MJ, Edgar JC, Ehrlichman RS, Mehta M, Roberts TP, Siegel SJ. Validating oscillations and delayed auditory responses as translational biomarkers of autism. Biol Psychiatry 2010;68:1100-1106.
- Kim JW, Seung H, Kwon KJ, Ko MJ, Lee EJ, Oh HA, Choi CS, Kim KC, Gonzales EL, You JS, Choi DH, Lee J, Han SH, Yang SM, Cheong JH, Shin CY, Bahn GH. Subchronic treatment of donepezil rescues impaired social, hyperactive, and stereotypic behavior in valproic acid-induced animal model of autism. PLoS One 2014;9:e104927.
- Roullet FI, Wollaston L, Decatanzaro D, Foster JA. Behavioral and molecular changes in the mouse in response to prenatal exposure to the anti-epileptic drug valproic acid. Neuroscience 2010;170:514-522.
- Portfors CV. Types and functions of ultrasonic vocalizations in laboratory rats and mice. J Am Assoc Lab Anim Sci 2007;46:28-34.
- Lewis MH, Bodfish JW. Repetitive behavior disorders in autism. Ment Retard Dev Disabil Res Rev 1998;4:80-89.
- Bodfish JW, Symons FJ, Parker DE, Lewis MH. Varieties of repetitive behavior in autism: comparisons to mental retardation. J Autism Dev Disord 2000;30:237-243.
- Campbell M, Locascio JJ, Choroco MC, Spencer EK, Malone RP, Kafantaris V, Overall JE. Stereotypies and tardive dyskinesia: abnormal movements in autistic children. Psychopharmacol Bull 1990;26:260-266.
- Prior M, Macmillan MB. Maintenance of sameness in children with Kanner's syndrome. J Autism Child Schizophr 1973;3:154-167.
- Langen M, Durston S, Kas MJ, van Engeland H, Staal WG. The neurobiology of repetitive behavior: …and men. Neurosci Biobehav Rev 2011;35:356-365.
- Lam KS, Bodfish JW, Piven J. Evidence for three subtypes of repetitive behavior in autism that differ in familiality and association with other symptoms. J Child Psychol Psychiatry 2008;49:1193-1200.
- Markram K, Rinaldi T, La Mendola D, Sandi C, Markram H. Abnormal fear conditioning and amygdala processing in an animal model of autism. Neuropsychopharmacology 2008;33:901-912.
- Zhang Y, Sun Y, Wang F, Wang Z, Peng Y, Li R. Downregulating the canonical Wnt/β-catenin signaling pathway attenuates the susceptibility to autism-like phenotypes by decreasing oxidative stress. Neurochem Res 2012;37:1409-1419.
- Mehta MV, Gandal MJ, Siegel SJ. mGluR5-antagonist mediated reversal of elevated stereotyped, repetitive behaviors in the VPA model of autism. PLoS One 2011;6:e26077.
- Argyropoulos A, Gilby KL, Hill-Yardin EL. Studying autism in rodent models: reconciling endophenotypes with comorbidities. Front Hum Neurosci 2013;7:417.
- Kwakye LD, Foss-Feig JH, Cascio CJ, Stone WL, Wallace MT. Altered auditory and multisensory temporal processing in autism spectrum disorders. Front Integr Nuerosci 2011;4:129.
- Woolfenden S, Sarkozy V, Ridley G, Coory M, Williams K. A systematic review of two outcomes in autism spectrum disorder - epilepsy and mortality. Dev Med Child Neurol 2012;54:306-312.
- Sobrian SK, Nandedkar AK. Prenatal antiepileptic drug exposure alters seizure susceptibility in rats. Pharmacol Biochem Behav 1986;24:1383-1391.
- Kim JW, Choi CS, Kim KC, Park JH, Seung H, Joo SH, Yang SM, Shin CY, Park SH. Gastrointestinal tract abnormalities induced by prenatal valproic Acid exposure in rat offspring. Toxicol Res 2013;29:173-179.
- Tsujino N, Nakatani Y, Seki Y, Nakasato A, Nakamura M, Sugawara M, Arita H. Abnormality of circadian rhythm accompanied by an increase in frontal cortex serotonin in animal model of autism. Neurosci Res 2007;57:289-295.
- Geyer MA, Markou A. In: Bloom FE, Kupfer DJ. Neuropsychopharmacology - 5th generation of progress. New York, NY: Raven Press, 1995; 1995. p. 787-798.
- Sarter M, Bruno JP. In: D'Haenen H, den Boer A, Willner P. Biological psychiatry. Chichester: John Wiley & Sons Ltd, 2002; 2002. p. 37-44.
- Buitelaar JK. Why have drug treatments been so disappointing?. Novartis Found Symp 2003;251:235-244.
- Nilsson M, Perfilieva E, Johansson U, Orwar O, Eriksson PS. Enriched environment increases neurogenesis in the adult rat dentate gyrus and improves spatial memory. J Neurobiol 1999;39:569-578.
- Rampon C, Tang YP, Goodhouse J, Shimizu E, Kyin M, Tsien JZ. Enrichment induces structural changes and recovery from nonspatial memory deficits in CA1 NMDAR1-knockout mice. Nat Neurosci 2000;3:238-244.
- Morley-Fletcher S, Rea M, Maccari S, Laviola G. Environmental enrichment during adolescence reverses the effects of prenatal stress on play behaviour and HPA axis reactivity in rats. Eur J Neurosci 2003;18:3367-3374.
- Schneider T, Turczak J, Przewłocki R. Environmental enrichment reverses behavioral alterations in rats prenatally exposed to valproic acid: issues for a therapeutic approach in autism. Neuropsychopharmacology 2006;31:36-46.
- Puurunen K, Sirviö J, Koistinaho J, Miettinen R, Haapalinna A, Riekkinen P, Sivenius J. Studies on the influence of enriched-environment housing combined with systemic administration of an alpha2-adrenergic antagonist on spatial learning and hyperactivity after global ischemia in rats. Stroke 1997;28:623-631.
- Johansson BB. Functional outcome in rats transferred to an enriched environment 15 days after focal brain ischemia. Stroke 1996;27:324-326.
- Fernández CI, Collazo J, Bauza Y, Castellanos MR, López O. Environmental enrichment-behavior-oxidative stress interactions in the aged rat: issues for a therapeutic approach in human aging. Ann N Y Acad Sci 2004;1019:53-57.
- Cotman CW, Berchtold NC. Exercise: a behavioral intervention to enhance brain health and plasticity. Trends Neurosci 2002;25:295-301.
- Krech D, Rosenzweig MR, Bennett EL. Effects of environmental complexity and training on brain chemistry. J Comp Physiol Psychol 1960;53:509-519.
- Ivinskis A, Homewood J. Effects of preweaning environmental enrichment on later problem-solving behavior in rats. Anim Learn Behav 1980;8:336-340.
- Sarter M, Bruno JP. Developmental origins of the age-related decline in cortical cholinergic function and associated cognitive abilities. Neurobiol Aging 2004;25:1127-1139.
- Barnes CA, Meltzer J, Houston F, Orr G, McGann K, Wenk GL. Chronic treatment of old rats with donepezil or galantamine: effects on memory, hippocampal plasticity and nicotinic receptors. Neuroscience 2000;99:17-23.
- Picciotto MR, Higley MJ, Mineur YS. Acetylcholine as a neuromodulator: cholinergic signaling shapes nervous system function and behavior. Neuron 2012;76:116-129.
- Picciotto MR, Higley MJ, Mineur YS. Acetylcholine as a neuromodulator: cholinergic signaling shapes nervous system function and behavior. Neuron 2012;76:116-129.
- Mikhail FM, Lose EJ, Robin NH, Descartes MD, Rutledge KD, Rutledge SL, Korf BR, Carroll AJ. Clinically relevant single gene or intragenic deletions encompassing critical neurodevelopmental genes in patients with developmental delay, mental retardation, and/or autism spectrum disorders. Am J Med Genet A 2011;155A:2386-2396.
- Petersen AK, Ahmad A, Shafiq M, Brown-Kipphut B, Fong CT, Anwar Iqbal M. Deletion 1q43 encompassing only CHRM3 in a patient with autistic disorder. Eur J Med Genet 2013;56:118-122.
- Kaminsky R, Moriarty TM, Bodine J, Wolf DE, Davidson M. Effect of famotidine on deficit symptoms of schizophrenia. Lancet 1990;335:1351-1352.
- Haas HL, Sergeeva OA, Selbach O. Histamine in the nervous system. Physiol Rev 2008;88:1183-1241.
- Shan L, Bossers K, Unmehopa U, Bao AM, Swaab DF. Alterations in the histaminergic system in Alzheimer's disease: a postmortem study. Neurobiol Aging 2012;33:2585-2598.
- Tiligada E, Kyriakidis K, Chazot PL, Passani MB. Histamine pharmacology and new CNS drug targets. CNS Neurosci Ther 2011;17:620-628.
- Linday LA. Oral famotidine: a potential treatment for children with autism. Med Hypotheses 1997;48:381-386.
- Rossi PG, Posar A, Parmeggiani A, Pipitone E, D'Agata M. Niaprazine in the treatment of autistic disorder. J Child Neurol 1999;14:547-550.
- Baronio D, Castro K, Gonchoroski T, de Melo GM, Nunes GD, Bambini-Junior V, Gottfried C, Riesgo R. Effects of an H3R antagonist on the animal model of autism induced by prenatal exposure to valproic acid. PLoS One 2015;10:e0116363.
- Bambini-Junior V, Zanatta G, Della Flora Nunes G, Mueller de Melo G, Michels M, Fontes-Dutra M, Nogueira Freire V, Riesgo R, Gottfried C. Resveratrol prevents social deficits in animal model of autism induced by valproic acid. Neurosci Lett 2014;583:176-181.
- Vang O, Ahmad N, Baile CA, Baur JA, Brown K, Csiszar A, Das DK, Delmas D, Gottfried C, Lin HY, Ma QY, Mukhopadhyay P, Nalini N, Pezzuto JM, Richard T, Shukla Y, Surh YJ, Szekeres T, Szkudelski T, Walle T, Wu JM. What is new for an old molecule? Systematic review and recommendations on the use of resveratrol. PLoS One 2011;6:e19881.
- Albani D, Polito L, Signorini A, Forloni G. Neuroprotective properties of resveratrol in different neurodegenerative disorders. Biofactors 2010;36:370-376.
- Maher P, Dargusch R, Bodai L, Gerard PE, Purcell JM, Marsh JL. ERK activation by the polyphenols fisetin and resveratrol provides neuroprotection in multiple models of Huntington's disease. Hum Mol Genet 2011;20:261-270.
- Sun AY, Wang Q, Simonyi A, Sun GY. Resveratrol as a therapeutic agent for neurodegenerative diseases. Mol Neurobiol 2010;41:375-383.
- Shaywitz BA, Yager RD, Klopper JH. Selective brain dopamine depletion in developing rats: an experimental model of minimal brain dysfunction. Science 1976;191:305-308.
- Choi CS, Hong M, Kim KC, Kim JW, Yang SM, Seung H, Ko MJ, Choi DH, You JS, Shin CY, Bahn GH. Effects of atomoxetine on hyper-locomotive activity of the prenatally valproate-exposed rat offspring. Biomol Ther 2014;22:406-413.
- Shin S, Jang JY, Park D, Yon JM, Baek IJ, Hwang BY, Nam SY, Yun YW, Kim KY, Joo SS, Kim YB. Korean red ginseng extract does not cause embryo-fetal death or abnormalities in mice. Birth Defects Res B Dev Reprod Toxicol 2010;89:78-85.
- Hwang SY, Kim WJ, Wee JJ, Choi JS, Kim SK. Panax ginseng improves survival and sperm quality in guinea pigs exposed to 2,3,7,8-tetrachlorodibenzo-p-dioxin. BJU Int 2004;94:663-668.
- Lee JH, Sul D, Oh E, Jung WW, Hwang KW, Hwang TS, Lee KC, Won NH. Panax ginseng effects on DNA damage, CYP1A1 expression and histopathological changes in testes of rats exposed to 2,3,7,8-tetrachlorodibenzo-p-dioxin. Food Chem Toxicol 2007;45:2237-2244.
- Kim P, Park JH, Kwon KJ, Kim KC, Kim HJ, Lee JM, Kim HY, Han SH, Shin CY. Effects of Korean red ginseng extracts on neural tube defects and impairment of social interaction induced by prenatal exposure to valproic acid. Food Chem Toxicol 2013;51:288-296.
- Tordjman S, Drapier D, Bonnot O, Graignic R, Fortes S, Cohen D, Millet B, Laurent C, Roubertoux PL. Animal models relevant to schizophrenia and autism: validity and limitations. Behav Genet 2007;37:61-78.
- Patterson PH. Modeling autistic features in animals. Pediatr Res 2011;69:34R-40R.