Articles

Article Tools
 View Full Text View Full Text |
 Abstract Abstract |
 Article as PDF Article as PDF |
 Print this Article Print this Article |
 Pubmed Pubmed |
 PMC PMC |
 PubReader PubReader |
 Export to Citation Export to Citation |
 Email Alerts Email Alerts |
 Open Access Open Access |
Share this article on :



Stats or Metrics
Article
Original Article
Exp Neurobiol 2019; 28(2): 270-278
Published online April 30, 2019
https://doi.org/10.5607/en.2019.28.2.270
© The Korean Society for Brain and Neural Sciences
Glutamine Supplementation Ameliorates Chronic Stress-induced Reductions in Glutamate and Glutamine Transporters in the Mouse Prefrontal Cortex
Ji Hyeong Baek†, Arul Vignesh†, Hyeonwi Son, Dong Hoon Lee, Gu Seob Roh, Sang Soo Kang, Gyeong Jae Cho, Wan Sung Choi, and Hyun Joon Kim*
Department of Anatomy and Convergence Medical Sciences, Institute of Health Sciences, Bio Anti-aging Medical Research Center, Gyeongsang National University School of Medicine, Jinju 52727, Korea.
Correspondence to: *To whom correspondence should be addressed.
TEL: 82-55-772-8034, FAX: 82-55-772-8039
e-mail: kimhj@gnu.ac.kr
†These authors contributed equally to this work.
Abstract
Chronic immobilization stress (CIS) induces low levels of glutamate (Glu) and glutamine (Gln) and hypoactive glutamatergic signaling in the mouse prefrontal cortex (PFC), which is closely related to the Glu-Gln cycle. A Gln-supplemented diet ameliorates CIS-induced deleterious changes. Here, we investigated the effects of CIS and Gln supplementation on Glu-Gln cycle-related proteins to characterize the underlying mechanisms. Using the CIS-induced depression mouse model, we examined the expression of 11 proteins involved in the Glu-Gln cycle in the PFC. CIS decreased levels of glutamate transporter 1 (GLT1) and sodium-coupled neutral amino acid transporter (SNAT) 1, SANT2, SNAT3, and SNAT5. Gln supplementation did not affect the non-stressed group but significantly increased GLT1 and SNATs of the stressed group. By immunohistochemical analysis, we confirmed that SNAT1 and SNAT2 were decreased in neurons and GLT1, SNAT3, and SNAT5 were decreased in astrocytes in the medial PFC of the stressed group, but Gln-supplemented diet ameliorated these decrements. Collectively, these results suggest that CIS may cause depressive-like behaviors by decreasing Glu and Gln transportation in the PFC and that a Gln-supplemented diet could prevent the deleterious effects of CIS.
Graphical Abstract
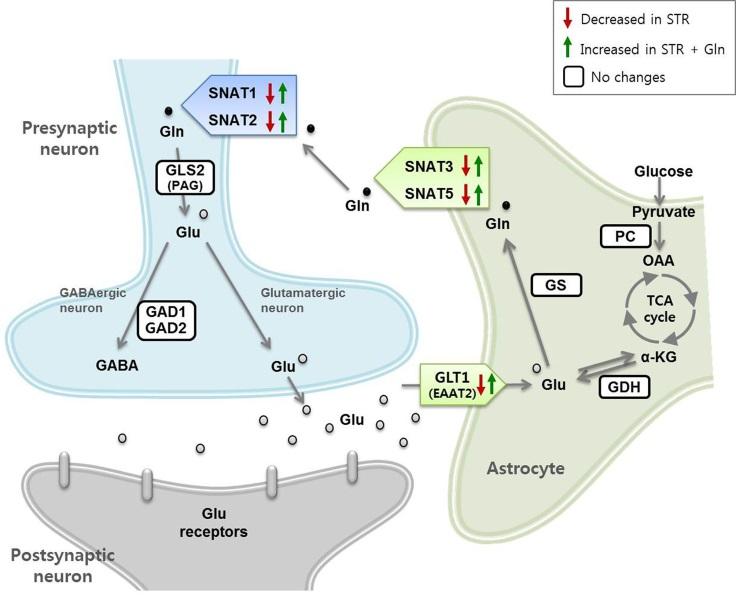
Keywords: Depressive disorder, Stress, Glutamate, Glutamine, Prefrontal cortex
INTRODUCTION
Glutamate (Glu) is a major excitatory neurotransmitter in the central nervous system (CNS) and is involved in the detoxification of ammonia and synthesis of peptides and proteins. Due to its multiple functions and extremely high intracellular concentration in the brain (1~10 mM), Glu must be tightly regulated to limit extracellular levels to ensure optimal neurotransmission and prevent potential excitotoxicity [1,2]. However, transporter-mediated uptake and recycling of Glu via the Glu-Glutamine (Glu-Gln) cycle is sensitive to stress and glucocorticoids. Emerging evidence has shown that disturbed glutamatergic neurotransmission in the CNS is the main cause for stress-induced psychiatric disorders including major depressive disorder (MDD) [1,3].
Astrocytes are the major glial cells and play a crucial role in regulating glutamatergic signaling via the Glu-Gln cycle [4,5,6,7,8]. The central functions of astrocytes include rapid elimination of Glu released from presynaptic neurons and mediating neurotransmitter metabolism via the Glu-Gln cycle, enabling astrocytes to control synaptic activity. However, stress and glucocorticoids (mainly corticosterone in rodents) increase extracellular Glu levels, thus disrupting optimal collaboration between astrocytes and neurons to produce desired functions [1,4].
A variety of studies in MDD patients and depressive rodent models have shown altered glutamatergic neurotransmission in the brain, such as low glutamatergic synaptic activity, reduced Glu and Gln levels, and changes in expression levels or activities of the Glu-Gln cycle proteins [3,9,10,11,12]. Disturbance of the Glu-Gln cycle via L-α-aminoadipic acid (an astrocyte-specific toxin), methionine sulfoximine (a Gln synthetase (GS) inhibitor), and α-methylamino-isobutyric acid (a blocker of neuronal Gln transporters) leads to depressive behaviors and decreased Glu and Gln levels in the prefrontal cortex (PFC) of rodents [3,13,14]. Moreover, direct infusion or supplementation of Gln reverses the depressive behaviors and Glu-Gln cycle impairments, implying that disrupted glutamatergic signaling and neuronal Gln deficiency mediate depression [3,12,14].
We recently demonstrated that chronic immobilization stress (CIS) causes depressive behaviors by reducing GS activity and Glu and Gln levels, ultimately leading to hypoactive glutamatergic neurotransmission in the medial PFC (mPFC) of mice; however, a Gln-supplemented diet reversed these effects [3]. To further investigate how CIS affected Glu and Gln levels and how exogenous Gln restored it, we evaluated changes in the expression levels of Glu-Gln cycle proteins in the PFC caused by CIS and Gln supplementation.
MATERIALS AND METHODS
Male 7-week-old C57BL/6 mice (Koatech, Pyeongtaek, Republic of Korea) were habituated for 1 week before experiments in a specific pathogen-free animal facility at the School of Medicine, Gyeongsang National University. Mice were individually housed at a constant temperature (22~24℃) under a 12-h light/dark cycle (lights on at 6 am) with free access to laboratory chow and water. Gln-supplemented diets (150 mg/kg) were fed to the Gln group, and calorie-balanced normal chow diets were fed to the control group (Uni Faith, Seoul, Republic of Korea) during the entire period of the experiments as previously described [3]. Mice were randomly grouped using a computer-generated list according to body weight. Control (CTL) and control-glutamine (CTL+Gln) groups were caged with or without Gln supplementation. Stress (STR) and stress-glutamine (STR+Gln) groups were exposed to stress with or without Gln supplementation. Animal use procedures were performed in accordance with the National Institutes of Health guidelines and an approved protocol (GNU-140225-M0012) by the Gyeongsang National University Institution Animal Care & Use Committee.
CIS was carried out as previously described [15]. Briefly, mice were repeatedly placed in a restrainer for 2 h/day (14:00~16:00) for 15 days under 200 lux light conditions. Body weight and food intake were measured every other day. After CIS, one batch of mice was subjected to depressive behavioral tests. The other batch was sacrificed by decapitation at 9:00~11:00 am and used for molecular analyses such as plasma corticosterone analysis and expression level analyses of Glu-Gln cycle proteins, without undergoing behavioral tests (Fig. 1A).
The sucrose preference test (SPT) was performed with some modifications as previously described to identify symptoms of anhedonia [14]. Briefly, mice were habituated for 48 h with a palatable sucrose solution (0.1 M), followed by a 24-h water deprivation period and a 6-h exposure to two identical bottles, one filled with sucrose solution and the other with water. Sucrose solution and water consumption were determined for 6 h. The sucrose preference was represented as the ratio of sucrose-to-water consumption.
The tail suspension test (TST) was conducted as previously described with some modifications [3]. Mice were individually suspended by the tail to a horizontal bar (the distance from the floor was 30 cm) using clear tape (distance from the tip of the tail was approximately 1 cm). The duration of immobility during the 6-min test session was recorded and analyzed by an animal behavior video analysis program (EthoVision; Noldus Information Technology, Wageningen, The Netherlands).
Mouse blood was collected in vacutainers containing K3EDTA. Plasma was isolated by centrifugation at 1,000×g, at 4℃ for 10 min. The samples were stored at −80℃ until use. Quantification of plasma corticosterone (CORT) levels was carried out using a CORT enzyme-linked immunosorbent assay kit (Cayman, Ann Arbor, MI, USA) according to the manufacturer's protocol.
Crude proteins were extracted from the PFC by homogenizing with glass beads and RIPA buffer (Elpis-Biotech, Daejeon, Republic of Korea) using a Bullet Blender (Next Advance, New York, NY, USA), followed by sonication for 2 min and centrifugation at 12,000×g at 4℃ for 10 min. Protein samples (10 µg) were separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis and transferred to polyvinylidene difluoride membranes. The membranes were blocked with 5% skim milk or 3% bovine serum albumin in Tris-buffered saline with Tween (50 mM Tris, 150 mM NaCl, 0.1% or 1% Tween-20, pH 7.4) for 2 h and incubated overnight at 4℃ with the following primary antibodies: PC (1:2,000, SC373937; Santa Cruz Biotechnology, Dallas, TX, USA), GDH [1:5,000, SC160383 (GDH1/2); Santa Cruz Biotechnology], GLT1 (1:1,000, SC7760; Santa Cruz Biotechnology), GS (1:10,000, MAB302; EMD Millipore, Temecula, CA, USA), SNAT3 (1:1,000, ab211516; Abcam, Cambridge, UK), SNAT5 (1:2,000, ab72717; Abcam), SNAT1 (1:1,000, SC67080; Santa Cruz Biotechnology) SNAT2 (1:1,000, SC166366; Santa Cruz Biotechnology), GLS2 (1:2,000, AB113509; Abcam), GAD1 (1:2,000, 5305; Cell Signaling, Danvers, MA, USA), GAD2 (1:2,000, 3988; Cell Signaling), α-tubulin (1:10,000, T5168; Sigma-Aldrich, St. Louis, MO, USA), and β-actin (1:10,000, A5441; Sigma-Aldrich). After secondary antibody binding for 2 h with anti-rabbit IgG-horseradish peroxidase (HRP) (1:10,000, NCI1460KR; Thermo Fisher Scientific, Waltham, MA, USA) or anti-mouse IgG-HRP (1:10,000, NCI1430KR; Thermo Fisher Scientific) antibodies, immunoblot signals were detected using an enhanced chemiluminescence detection kit (Thermo Fisher Scientific) and iBright FL1000 (Thermo Fisher Scientific). Band intensities were calculated using iBright analysis software (Thermo Fisher Scientific). Expression levels were normalized against the internal controls, α-tubulin or β-actin.
IHC for GLT1 and SNATs were performed as previously described [3] with some modifications. Twenty-four hours after the last stress, mice were deeply anesthetized with avertin and perfused with phosphate-buffered saline (pH 7.4) and 4% paraformaldehyde. The brains were collected, postfixed, sectioned at 40-µm thickness, and incubated with an antibody (1:20~1:200) at room temperature or 4℃ overnight. An anti-glial fibrillary acidic protein (GFAP) antibody (Z0334) (1:200; Dako, Glostrup, Denmark), anti-GS antibody (MAB302) (1:200; Merck Millipore, MA, USA), anti-NeuN antibody (MAB377) (1:200; Merck Millipore), and anti-GLS2 (AB113509) (1:200; Abcam) were used as cellular distribution markers. The slices were then incubated with Alexa Fluor 594- and 488-conjugated secondary antibodies (1:1,000; Invitrogen, Carlsbad, CA, USA). Digital images were captured using a spinning disk confocal microscope equipped with an Olympus Disk Spinning Unit (Olympus, Tokyo, Japan) and analyzed by ImageJ software (NIH).
Data are presented as means± SEMs. Student's
RESULTS
CIS decreased the body weight of the stress group (STR) and Gln-supplemented STR group (STR+Gln), but there was no difference in the food intake between the tested groups (Fig. 1B and 1C). Gln supplementation did not affect body weight or food intake.
The STR group showed higher immobility in the TST and less sucrose preference compared with the CTL group (Fig. 1D and 1E), indicating that CIS induced depressive behaviors. Gln prevented these in the STR group (STR vs. STR+Gln), but no significant effect was observed in normal control mice (CTL vs. CTL+Gln). Moreover, basal plasma CORT levels also markedly increased after CIS, but they remained at the CTL level with Gln supplementation (Fig. 1F).
The effects of CIS and Gln on Glu-Gln cycle protein expression in the PFC were investigated by immunoblotting (Fig. 2). The expression level of the Glu transporter GLT1 decreased after CIS. In addition, the Gln transporters SNAT2, SNAT3, and SNAT5 decreased in the PFC of CIS-subjected mice. Gln supplementation did not affect the non-stressed group (CTL+Gln). However, the expression levels of SNAT1, SNAT2, SNAT3, and SNAT5 in the STR+Gln group were significantly higher than those of the STR group (Fig. 2).
A mature glycosylated form (~60 kDa) and an immature form (non- or partially glycosylated, ~50 kDa) of SNAT2 [16] showed different expression patterns in response to CIS; the mature form decreased, whereas the immature form increased. The mature and immature forms of the other Gln transporters (SNAT1, SNAT3, and SNAT5) showed similar expression patterns (Fig. 2).
To confirm the immunoblot results and cellular locations, expression level changes of GLT1 and SNATs in response CIS and Gln supplementation were analyzed by IHC in the mPFC (Fig. 3). Double-positive cells for each protein with neuronal or astrocytic marker were decreased by CIS, but not in the stressed group with Gln supplementation, supporting the immunoblot results. In addition, IHC showed a significant GLT1 induction by Gln (Fig. 3A) and SNAT1 reduction in the mPFC by CIS (Fig. 3B). These expressional changes were not significant in the immunoblots performed with crude PFC protein samples.
To assess whether CORT levels contribute to the regulation of Glu-Gln cycle proteins, we analyzed the correlation between plasma CORT and the Glu-Gln cycle protein expression levels in response to CIS (Fig. 4). GLT1 and mature and immature SNAT2 expression levels strongly correlated with plasma CORT levels (p-values near or <0.01). In addition, SNAT5 also showed a negative correlation with plasma CORT levels with a p-value of approximately 0.05, suggesting that the expression of GLT1, SNAT2, and SNAT5 might be associated with plasma CORT levels. SNAT1 and SNAT3 had weak or insignificant correlations with plasma CORT levels.
DISCUSSION
Reduced levels of Glu and Gln in the frontal cortex have been consistently reported in MDD patients [17,18,19]. In the brain, Gln is exclusively synthesized in astrocytes by GS and is involved in the Glu-Gln cycle to maintain glutamatergic neurotransmissions [20]. Our recent study showed that CIS reduced GS activity, Glu-Gln levels, and the spontaneous excitatory synaptic current (sEPSC) frequency of glutamatergic neurons in the mPFC, which was resulted in depressive behaviors [3]. Moreover, we showed that Gln supplementation prevented these deleterious changes induced by CIS, implying an antidepressant effect of Gln [3]. In the present study, we investigated the effects of CIS and Gln supplementation on Glu-Gln cycle-related protein levels to identify the underlying mechanisms that could cause Glu and Gln shortages in the PFC of a CIS-induced depression mouse model.
The effects of CIS and Gln on behavior and plasma CORT levels were similar to our previous studies [3,14,15], confirming that CIS induces depressive behaviors in mice and Gln supplementation prevents chronic stress-induced depression (Fig. 1). We found that GLT1, SNAT1, SNAT2, SNAT3, and SNAT5 were decreased in the PFC after CIS (Fig. 2, 3, and 5). However, Gln supplementation increased their levels under the stress condition. These results suggest that CIS disturbed the Glu-Gln cycle, possibly resulting in a shortage of Glu and Gln levels in astrocytes and neurons, whereas Gln supplementation ameliorated deleterious CIS-induced changes in the Glu-Gln cycle by facilitating translocation of Glu into astrocytes and Gln into neurons for glutamatergic signaling.
Previous studies have suggested low GLT1 in the brain (anterior cingulate cortex, left dorsolateral PFC, amygdala, and hippocampus) as a possible cause of depressive disorders [11,21,22,23,24]. Blockade of GLT1 induced anhedonia and impaired spatial memory [25], whereas stimulation of GLT1 expression had antidepressant effects [26,27,28,29,30]. The decrease of astrocytic Glu uptake due to GLT1 reduction might lead to a shortage of Gln that is synthesized from Glu by GS in astrocytes.
However, Glu in the synaptic cleft would increase, at least at the early stage of GLT1 reduction, because Glu removal decreased. GLT1 is the predominant Glu transporter in the mature brain and accounts for ~95% of total Glu uptake in the forebrain [7,31]. The reduction of GLT1 is therefore potentially neurotoxic and could affect glutamatergic signaling efficiency [11].
We reported that specific blocking of neuronal glutamine transportation mediated by SNAT1/2 caused depressive-like behaviors with low levels of Glu-Gln [14] and disturbed glutamatergic sEPSCs in response to exogenous Glu and Gln [3]. In the present study, we found decreased SNAT1/2 in the STR and increments in the STR+Gln group (Fig. 2 and 3). These findings suggest that neuronal Gln transportation might be a crucial step for normal glutamatergic signaling and maintenance of normal behaviors during chronic stress, which also support our previous hypotheses [3,14]. The functions of SNAT3 and SNAT5 in MDD pathogenesis remain unclear. The meaning of those proteins' changes by CIS and Gln supplementation revealed by this study would deserve further studies in relation with etiology of chronic stress-induced depression and development for new antidepressants.
High molecular-weight (~60 kDa) SNAT2 decreased, whereas 50-kDa SNAT2 accumulated in the PFC following CIS. It was previously reported that the 60-kDa band is a fully glycosylated mature cell-surface transporter, while the lighter band is a partially processed immature intracellular SNAT2 [16]. The decrease of mature SNAT2 and increase of immature SNAT2 by CIS could be due to the blocking or delay of normal protein processing. The other SNATs (SNAT1, SNAT3, and SNAT5) did not show any significant difference in expression between the mature and immature forms (Fig. 2), which means that the protein processing pathway for SNAT2 might have been more sensitive to CIS compared to the other SNATs.
To our knowledge, no studies have examined expressional changes of GLT1 and SNATs relative to plasma CORT levels in the brain. Our results showed that GLT1, SNAT2, and SNAT5 expression correlated with plasma CORT levels (Fig. 4). The relationship between CORT and these transporters should be further studied to clarify the precise regulatory mechanism of GLT1, SNAT2, and SNAT5 expression and their roles in the etiology of depression.
In summary, the results showed that CIS disturbed the Glu-Gln cycle in the PFC. To the best of our knowledge, this is the first study to investigate expressional changes of Glu-Gln cycle proteins as a whole in response to chronic stress. Our findings could help identify new antidepressant targets related to chronic stress. In addition, the results demonstrate that Gln supplementation facilitates normal Glu-Gln cycling under stressful conditions, consistent with our previous studies demonstrating the antidepressant potential of Gln [3,14]. In conclusion, CIS decreased Glu and Gln transporter levels, and Gln supplementation increased their expression in the PFC, suggesting that Glu and Gln transporters have important roles in depression pathogenesis and the antidepressant effect of Gln. However, to confirm our result that Gln is a potent antidepressant, the effects of Gln supplementation after CIS, but not during CIS, should be evaluated in further studies.
Figures

{kind=link}

{kind=link}

{kind=link}

{kind=link}

{kind=link}
References
- Popoli M, Yan Z, McEwen BS, Sanacora G. The stressed synapse: the impact of stress and glucocorticoids on glutamate transmission. Nat Rev Neurosci 2011;13:22-37.

- Danbolt NC. Glutamate uptake. Prog Neurobiol 2001;65:1-105.
- Son H, Baek JH, Go BS, Jung DH, Sontakke SB, Chung HJ, Lee DH, Roh GS, Kang SS, Cho GJ, Choi WS, Lee DK, Kim HJ. Glutamine has antidepressive effects through increments of glutamate and glutamine levels and glutamatergic activity in the medial prefrontal cortex. Neuropharmacology 2018;143:143-152.
- Ransom BR, Ransom CB. Astrocytes: multitalented stars of the central nervous system. Methods Mol Biol 2012;814:3-7.
- Bélanger M, Allaman I, Magistretti PJ. Brain energy metabolism: focus on astrocyte-neuron metabolic cooperation. Cell Metab 2011;14:724-738.
- Botman D, Tigchelaar W, Van Noorden CJ. Determination of glutamate dehydrogenase activity and its kinetics in mouse tissues using metabolic mapping (quantitative enzyme histochemistry). J Histochem Cytochem 2014;62:802-812.
- Haugeto O, Ullensvang K, Levy LM, Chaudhry FA, Honoré T, Nielsen M, Lehre KP, Danbolt NC. Brain glutamate transporter proteins form homomultimers. J Biol Chem 1996;271:27715-27722.
- Miller KE, Richards BA, Kriebel RM. Glutamine-, glutamine synthetase-, glutamate dehydrogenase- and pyruvate carboxylase-immunoreactivities in the rat dorsal root ganglion and peripheral nerve. Brain Res 2002;945:202-211.
- Abdallah CG, Jiang L, De Feyter HM, Fasula M, Krystal JH, Rothman DL, Mason GF, Sanacora G. Glutamate metabolism in major depressive disorder. Am J Psychiatry 2014;171:1320-1327.
- Bernard R, Kerman IA, Thompson RC, Jones EG, Bunney WE, Barchas JD, Schatzberg AF, Myers RM, Akil H, Watson SJ. Altered expression of glutamate signaling, growth factor, and glia genes in the locus coeruleus of patients with major depression. Mol Psychiatry 2011;16:634-646.
- Choudary PV, Molnar M, Evans SJ, Tomita H, Li JZ, Vawter MP, Myers RM, Bunney WE, Akil H, Watson SJ, Jones EG. Altered cortical glutamatergic and GABAergic signal transmission with glial involvement in depression. Proc Natl Acad Sci U S A 2005;102:15653-15658.
- Son H, Jung S, Shin JH, Kang MJ, Kim HJ. Anti-stress and anti-depressive effects of spinach extracts on a chronic stress-induced depression mouse model through lowering blood corticosterone and increasing brain glutamate and glutamine levels. J Clin Med 2018;7:406.
- Banasr M, Duman RS. Glial loss in the prefrontal cortex is sufficient to induce depressive-like behaviors. Biol Psychiatry 2008;64:863-870.
- Lee Y, Son H, Kim G, Kim S, Lee DH, Roh GS, Kang SS, Cho GJ, Choi WS, Kim HJ. Glutamine deficiency in the prefrontal cortex increases depressive-like behaviours in male mice. J Psychiatry Neurosci 2013;38:183-191.
- Joo Y, Choi KM, Lee YH, Kim G, Lee DH, Roh GS, Kang SS, Cho GJ, Choi WS, Kim HJ. Chronic immobilization stress induces anxiety- and depression-like behaviors and decreases transthyretin in the mouse cortex. Neurosci Lett 2009;461:121-125.
- Nardi F, Hoffmann TM, Stretton C, Cwiklinski E, Taylor PM, Hundal HS. Proteasomal modulation of cellular SNAT2 (SLC38A2) abundance and function by unsaturated fatty acid availability. J Biol Chem 2015;290:8173-8184.
- Arnone D, Mumuni AN, Jauhar S, Condon B, Cavanagh J. Indirect evidence of selective glial involvement in glutamate-based mechanisms of mood regulation in depression: meta-analysis of absolute prefrontal neuro-metabolic concentrations. Eur Neuropsychopharmacol 2015;25:1109-1117.
- Luykx JJ, Laban KG, van den Heuvel MP, Boks MP, Mandl RC, Kahn RS, Bakker SC. Region and state specific glutamate downregulation in major depressive disorder: a meta-analysis of 1H-MRS findings. Neurosci Biobehav Rev 2012;36:198-205.
- Taylor MJ. Could glutamate spectroscopy differentiate bipolar depression from unipolar?. J Affect Disord 2014;167:80-84.
- Hertz L, Zielke HR. Astrocytic control of glutamatergic activity: astrocytes as stars of the show. Trends Neurosci 2004;27:735-743.
- Moghaddam B, Adams B, Verma A, Daly D. Activation of glutamatergic neurotransmission by ketamine: a novel step in the pathway from NMDA receptor blockade to dopaminergic and cognitive disruptions associated with the prefrontal cortex. J Neurosci 1997;17:2921-2927.
- Valentine GW, Sanacora G. Targeting glial physiology and glutamate cycling in the treatment of depression. Biochem Pharmacol 2009;78:431-439.
- John CS, Sypek EI, Carlezon WA, Cohen BM, Öngür D, Bechtholt AJ. Blockade of the GLT-1 transporter in the central nucleus of the amygdala induces both anxiety and depressive-like symptoms. Neuropsychopharmacology 2015;40:1700-1708.
- Gourley SL, Espitia JW, Sanacora G, Taylor JR. Antidepressant-like properties of oral riluzole and utility of incentive disengagement models of depression in mice. Psychopharmacology (Berl) 2012;219:805-814.
- Bechtholt-Gompf AJ, Walther HV, Adams MA, Carlezon WA, Ongür D, Cohen BM. Blockade of astrocytic glutamate uptake in rats induces signs of anhedonia and impaired spatial memory. Neuropsychopharmacology 2010;35:2049-2059.
- Mineur YS, Picciotto MR, Sanacora G. Antidepressant-like effects of ceftriaxone in male C57BL/6J mice. Biol Psychiatry 2007;61:250-252.
- Sanacora G, Kendell SF, Levin Y, Simen AA, Fenton LR, Coric V, Krystal JH. Preliminary evidence of riluzole efficacy in antidepressant-treated patients with residual depressive symptoms. Biol Psychiatry 2007;61:822-825.
- Banasr M, Chowdhury GM, Terwilliger R, Newton SS, Duman RS, Behar KL, Sanacora G. Glial pathology in an animal model of depression: reversal of stress-induced cellular, metabolic and behavioral deficits by the glutamate-modulating drug riluzole. Mol Psychiatry 2010;15:501-511.
- Rothstein JD, Patel S, Regan MR, Haenggeli C, Huang YH, Bergles DE, Jin L, Dykes Hoberg M, Vidensky S, Chung DS, Toan SV, Bruijn LI, Su ZZ, Gupta P, Fisher PB. Beta-lactam antibiotics offer neuroprotection by increasing glutamate transporter expression. Nature 2005;433:73-77.
- Ding XF, Li YH, Chen JX, Sun LJ, Jiao HY, Wang XX, Zhou Y. Involvement of the glutamate/glutamine cycle and glutamate transporter GLT-1 in antidepressant-like effects of Xiao Yao san on chronically stressed mice. BMC Complement Altern Med 2017;17:326.
- Tanaka K, Watase K, Manabe T, Yamada K, Watanabe M, Takahashi K, Iwama H, Nishikawa T, Ichihara N, Kikuchi T, Okuyama S, Kawashima N, Hori S, Takimoto M, Wada K. Epilepsy and exacerbation of brain injury in mice lacking the glutamate transporter GLT-1. Science 1997;276:1699-1702.