Articles

Article Tools
 View Full Text View Full Text |
 Abstract Abstract |
 Article as PDF Article as PDF |
 Print this Article Print this Article |
 Pubmed Pubmed |
 PMC PMC |
 PubReader PubReader |
 Export to Citation Export to Citation |
 Email Alerts Email Alerts |
 Open Access Open Access |
Share this article on :



Stats or Metrics
Article
Original Article
Exp Neurobiol 2019; 28(3): 362-375
Published online June 26, 2019
https://doi.org/10.5607/en.2019.28.3.362
© The Korean Society for Brain and Neural Sciences
Quantitative Proteomic Analysis Reveals Impaired Axonal Guidance Signaling in Human Postmortem Brain Tissues of Chronic Traumatic Encephalopathy
Baibin Bi1,2,3,†, Han-Pil Choi4,†, Seung Jae Hyeon5,†, Shengnan Sun2, Ning Su1, Yuguang Liu3, Junghee Lee1,4,6, Neil W. Kowall1,4,6, Ann C. McKee1,4,6*, Jing-Hua Yang1,2,4*, and Hoon Ryu1,4,5,6*
1Departments of Neurology, Pathology, and Surgery, Boston University School of Medicine, Boston, MA 02118, USA.
2Cancer Research Center, Shandong University School of Medicine, Jinan 250012, China.
3Department of Neurosurgery, Qilu Hospital of Shandong University, Jinan 250012, China.
4Proteomics Laboratory, VA Boston Healthcare System, Boston, MA 02130, USA.
5Center for Neuromedicine, Brain Science Institute, Korea Institute of Science and Technology, Seoul 04535, Korea.
6Boston University Alzheimer's Disease Center (BU ADC) and Chronic Traumatic Encephalopathy (CTE) Center, Boston University School of Medicine, Boston, MA 02118, USA.
Correspondence to: *To whom correspondence should be addressed.
Ann C. McKee, TEL: 1-857-364-5707, FAX: 1-857-364-4540, e-mail: amckee@bu.edu
Jing-Hua Yang, TEL: 1-857-364-5611, FAX: 1-857-364-5627, e-mail: jyang@bu.edu
Hoon Ryu, TEL: 1-857-364-5910, FAX: 1-857-364-4540, e-mail: hoonryu@bu.edu
†These authors are equally contributed.
Abstract
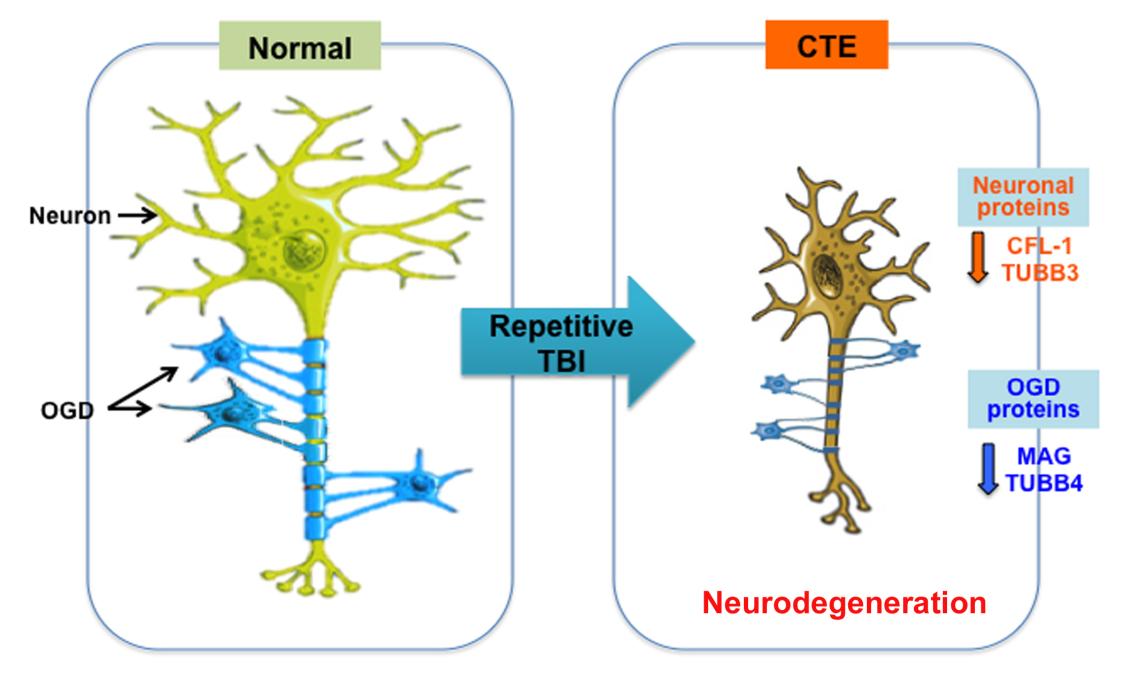
Chronic traumatic encephalopathy (CTE) is a distinct neurodegenerative disease that associated with repetitive head trauma. CTE is neuropathologically defined by the perivascular accumulation of abnormally phosphorylated tau protein in the depths of the sulci in the cerebral cortices. In advanced CTE, hyperphosphorylated tau protein deposits are found in widespread regions of brain, however the mechanisms of the progressive neurodegeneration in CTE are not fully understood. In order to identify which proteomic signatures are associated with CTE, we prepared RIPA-soluble fractions and performed quantitative proteomic analysis of postmortem brain tissue from individuals neuropathologically diagnosed with CTE. We found that axonal guidance signaling pathwayrelated proteins were most significantly decreased in CTE. Immunohistochemistry and Western blot analysis showed that axonal signaling pathway-related proteins were down regulated in neurons and oligodendrocytes and neuron-specific cytoskeletal proteins such as TUBB3 and CFL1 were reduced in the neuropils and cell body in CTE. Moreover, oligodendrocyte-specific proteins such as MAG and TUBB4 were decreased in the neuropils in both gray matter and white matter in CTE, which correlated with the degree of axonal injury and degeneration. Our findings indicate that deregulation of axonal guidance proteins in neurons and oligodendrocytes is associated with the neuropathology in CTE. Together, altered axonal guidance proteins may be potential pathological markers for CTE.
Graphical Abstract
Keywords: Chronic traumatic encephalopathy (CTE), Quantitative proteomics, Axonal guidance, Oligodendrocyte, Neuron, Neurodegeneration
INTRODUCTION
Traumatic brain injury (TBI) is a leading cause of morbidity and mortality, and over 1.7 M individuals suffer from TBI in the US every year [1,2,3]. TBI frequently occurs in sports, car accidents, and falls. Annual healthcare costs of TBI are estimated to be over $60 billion in the US [3]. Notably, it has been reported that single and repetitive TBI are associated with the development of neurodegenerative disorders, including chronic traumatic encephalopathy (CTE) and Alzheimer's disease (AD) [4,5,6,7,8,9,10,11,12,13,14,15].
CTE has been neuropathologically confirmed in contact sport athletes including football players [11,12,13,14,15,16,17] and military veterans exposed to blast [14,18]. The pathological criteria for the diagnosis of CTE were proposed by McKee and colleagues and confirmed by a NIH consensus panel of expert neuropathologists [14,17]. The severity of hyperphosphorylated tau pathology in CTE has been divided into 4 stages (Stages I thorough IV) that are significantly associated with increased age and length of a football playing career [14]. Other neuropathological features of CTE include axonal injury [14], neuroinflammation [19,20] and TDP43 deposition [14,21]. Individuals with CTE show clinical symptoms of behavior and mood dysfunction including aggression, depression, impulsivity, and suicidality, and cognitive changes of memory loss and executive dysfunction [14,16,22]. The symptoms of CTE often develop after a latent period of several years to even decades after the trauma has occurred [13].
Interestingly, CTE-like neuropathology has been observed in acute closed head injury such as concussion, subconcussion and blast injury [18,23]. In the blast neurotrauma, the axonal injury, blood brain barrier dysfunction, astrocytosis, microgliosis and hyperphosphorylated tau pathology are found as similar to CTE [18,23]. Together, it has been proposed that head trauma-induced axonal injury is directly linked to the development of neurodegenerative pathology in both the acute and the repetitive head injury [24,25,26,27,28,29,30,31].
In the current study, we performed mass spectrometry-based proteomic analysis of post-mortem brain tissue from individuals neuropathologically diagnosed with CTE and normal subjects in order to identify and characterize the protein signatures associated with CTE. We further analyzed significantly altered protein clusters by ingenuity pathway analysis (IPA). We discovered that axonal guidance signaling pathway-related proteins are most significantly affected in CTE. Western blot analysis and immunohistochemistry verified that neuron-specific cytoskeletal proteins (such as CFL-1 and TUBB3) and oligodendrocyte-specific proteins (such as MAG and TUBB4) were down regulated in both gray matter and white matter in CTE. Our data indicate that alteration of axonal guidance signaling proteins is closely linked to the neuronal and oligodendrocyte pathology found in CTE.
MATERIALS AND METHODS
Neuropathological processing of control and CTE human brain samples followed the procedures previously established for the Boston University Alzheimer's Disease Center (BUADC) and Chronic Traumatic Encephalopathy (CTE) Center. Institutional review board approval for ethical permission was obtained through the BUADC and CTE Center and Boston University School of Medicine. Next of kin provided informed consent for brain donation. The study was performed in accordance with institutional regulatory guidelines and principles of human subject protection in the Declaration of Helsinki. Detailed information of brain tissues is described in Supplementary Table 1.
Proteomic analysis involved (1) frozen tissue collection, (2) protein extraction, (3) sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) protein fractionation, (4) in-gel tryptic digestion and desalting, (5) LC-MS/MS, (6) label-free quantification, and (7) pathway and interaction network analyses and confirmation with Western blotting and immunohistochemistry staining (Fig. 1A and 1B). Frozen normal (n=3) and CTE (n=3) postmortem brain tissue (superior frontal cortex) samples were minced and homogenized in RIPA buffer (50 mM Tris-Cl pH 8.0, 150 mM NaCl, 1% NP-40, 0.5% sodium deoxycholate, 0.1% SDS) with protease inhibitor cocktail and phosphatase inhibitor cocktail. The tissue lysates were centrifuged, and the supernatants were collected and stored at −80℃until analysis. Protein concentration was determined with BioRad protein assay according to the manufacturer's instructions.
The proteins were fractionated using one-dimensional sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE). About 100 µg of proteins from normal (3 independent) (n=3) and CTE brain (3 independent) (n=3) tissues were separated on SDS-PAGE, roughly divided into 10 bands according to molecular weights, and digested, in-gel, with trypsin except reduction and alkylation steps [32]. The reduction and alkylation steps were omitted to avoid unwanted protein modification by dithiothreitol (DTT) and iodoacetamide. Instead, the tryptic digestion time was extended together with additional trypsin treatment to ensure complete digestion. The extracted peptides from each gel band were desalted by ZipTip (Millipore, Billerica, MA) and subjected to peptide fractionation using an EASY-nLC 1000 system (Thermo Scientific) equipped with a long C18 column (300×Ø 0.075 mm, C18 resin with 3 µm particle size, 100 Å pore diameter, Reprosil, Germany). Samples were analyzed using 120 min linear gradient of 5~35% acetonitrile/0.1% formic acid at a flow rate of 300 nL/min. The MS and MS/MS spectra were acquired by a LTQ-Orbitrap Elite mass spectrometer (Thermo Scientific) in a datadependent mode, in which MS/MS fragmentation with normalized collision energy of 35% was performed using the 30 most intense peaks above a threshold ion count of 1,000 in every full MS scan. Full MS spectra of 350~1,800
The acquired MS/MS spectra were searched against the human protein database (UniProtKB; 72277 entries) using the Proteome Discoverer 1.4 software (Thermo Scientific) with the built-in MASCOT and SEQUEST search engine. Trypsin (full cleavage) was specified as cleavage enzyme allowing up to two missing cleavages. Mass tolerance was set to 10 ppm for precursor ions and 0.5 Da for fragment ions. Methionine oxidation was selected as a dynamic modification, and the false discovery rate (FDR) was 1%.
Progenesis LC-MS software (version 4.1; Nonlinear Dynamics, UK) was employed for label-free quantification. The acquired spectra (raw data) of every fraction from CTE and control brain tissues were input, transformed and stored as peak lists with abundance and
For pathway analysis, Ingenuity Pathways Analysis (IPA, Ingenuity Systems) software was employed. It evaluates the association between differentially expressed proteins and pathways using p-value. The smaller the p-value is, the tighter the association is there. p<0.05 was considered statistically significant. Interaction network analysis was performed using the Search Tool for the Retrieval of Interacting Genes (STRING) 10.0 database (http://string-db.org) [33]. Medium confidence (0.4000) were used and active interaction sources included all default settings, such as text mining, experiments, database, co-expression, neighborhood, gene fusion, co-occurrence. STRING analysis options were based on ‘evidence’ mode. Disconnected nodes were hidden and no proteins were removed or added. Heat map presentation of the differentially expressed proteins was generated with Heml (Heatmap Illustrator) [34]. Color scale was based on the log2 transformed expression values of proteins in CTE or control. Otherwise, KEGG (Kyoto Encyclopedia of Genes and Genomes) pathway of up-regulated protein signatures was analyzed by DAVID (6.8 version).
Western blot was performed as previously described [35]. Twenty µg of protein was subjected to SDS-PAGE (13%) and blotted with anti-TUBB3 (05-559, Millipore.), anti-CFL1 (sc-53934, SantaCruz Biotech.), anti-TUBB4 and anti-MAG (ab179504 and ab89780, Abcam.) antibody. Protein loading was controlled by probing for GAPDH (AP0066, Bioworld Technology.) on the same membrane.
Immunohistochemical detection of tubulin βIII (TUBB3) and myelin associated glycoprotein (MAG) was performed on paraffin sections of the cortex tissue from fixed normal and CTE postmortem brains as previously described [36]. Briefly, paraffin-embedded sections were deparaffinized, rehydrated and treated with 3% H2O2 for antigen retrieval. After blocked with TBS-T containing 5% fetal bovine serum for 1 hour, sections were then incubated overnight in TBS-T with 5% fetal bovine serum and mouse anti-TUBB3 (1:100; 05-559, Millipore), mouse anti-MAG (1:100; ab89780, Abcam). The following morning, sections were rinsed three times in TBS-T, followed by a 2~3 hour incubation in TBS-T containing a goat anti-mouse peroxidase-conjugated secondary antibody to detect TUBB3, MAG respectively. The sections were then rinsed three times in TBS-T and further incubated in TBS-T with ABC solution (Vector Kit) for 1 hour. Antibody complexes were visualized by using diaminobenzidine (DAB) and analyzed by a bright field microscopy at various magnifications.
Data were analyzed using SigmaPlot and represented as the mean±standard error of the mean (SEM). For single comparison, the significance of differences between means was assessed by Student's
RESULTS
In the first series of experiments, frozen cortex (superior frontal) tissue lysates from the normal (3 independent) and CTE postmortem brains (3 independent) were pre-fractionated separately by SDS-PAGE to yield 10 fractions according to their molecular weights. After in-gel tryptic digestion of each fraction, LC-MS/MS followed by MASCOT and SEQUEST search engine yielded 3591, 3582 and 3635 proteins for the CTE brains and 3909, 3921 and 3920 proteins for the controls with 1% FDR from three replicates, respectively. In combination, a total of 4695 proteins in CTE brains and 5057 proteins in normal controls were identified. In total, 6218 non-redundant proteins were identified from the CTE and normal control brains (Fig. 1C and Supplementary Table 2).
Among the identified proteins, ion intensity-based label-free quantification approach using Progenesis LC-MS software revealed 1558 up-regulated proteins and 1837 down-regulated proteins in the CTE brains versus controls with p<0.05. The final panel of altered proteins in CTE was narrowed down by selecting the proteins which had at least 2 unique peptides for quantification and had expression levels with at least 1.5-fold of increase or decrease, resulting in 486 up-regulated and 885 down-regulated proteins in comparison with controls (Fig. 1D and Supplementary Table 3).
To get better understanding of the 1371 altered proteins (486 upregulated and 885 down-regulated with at least 1.5-fold changes) in CTE, IPA was employed to explore pathway property. IPA analysis revealed axonal guidance signaling was the top pathway which was the most highly associated with the differentially expressed proteins in CTE (Fig. 2A). Moreover, the highest number of proteins among the CTE-associated proteins was associated with axonal guidance signaling (Supplementary Table 4).
Interaction network analysis with STRING revealed that multiple interaction network and interaction clusters were formed between the 1371 CTE-associated proteins (Fig. 2B). When down-regulated proteins associated with axonal guidance signaling (the top pathway in IPA analysis) were applied, interaction network analysis revealed the following clusters were formed between those down-regulated proteins associated with axonal guidance signaling: cytoskeletal protein, myelin sheath related protein, protein phosphatase related protein, protein kinase-related protein, kinase signaling-related protein, G-coupled protein and Rho GTPase-related protein (Fig. 2C).
In order to confirm the change of axonal guidance signalingrelated protein levels in CTE, we performed Western blot and analyzed the protein levels semiquantitatively by densitometry (Fig. 3). The protein levels of neuronal markers such as cofilin-1 (CFL-1) and tubulin βIII (TUBB3) were significantly reduced in the cortex of CTE compared to normal (Fig. 3A). In addition, the protein levels of oligodendrocyte markers such as myelin associated glycoprotein (MAG) and tubulin βIV (TUBB4) were significantly reduced in the cortex of CTE compared to normal (Fig. 3B). On the other hand, the levels of Rho GTPase family proteins such as Mitogen-Activated Protein Kinase Kinase 2 (MEK2/MAP2K2) and Ras Homolog Family Member G (RHOG) were significantly reduced in the cortex of CTE compared to normal (Fig. 3C).
Because several axonal guidance signaling-related proteins are expressed in a cell type-specific manner, we further performed immunocytochemistry (IHC) and determined the localization of TUBB3 and MAG in the cortex of normal and CTE brain tissues (Fig. 4). As expected, the immunoreactivity of TUBB3, a neuronal cytoskeletal marker, was markedly decreased in the cell body and the dendrite of pyramidal neurons in the grey matter of CTE cortex compared to the normal brain (Fig. 4A, left panels). The immunoreactivity of TUBB3 was also reduced in the white matter of CTE cortex compared to the normal brain (Fig. 4A, right panels). Densitometry analysis of IHC data confirmed that TUBB3 protein level was significantly reduced in both the white and the gray matter of CTE patients (Fig. 4B). In addition, the immunoreactivity of MAG, an axonal and oligodendrocyte marker, was markedly reduced in the neurites of the grey matter of CTE cortex compared to the normal brain (Fig. 4C, left panel). The immunoreactivity of MAG was markedly decreased in the white matter CTE cortex as well (Fig. 4C, right panel). Densitometry analysis of IHC data confirmed that MAG protein level was significantly reduced in both the white and the gray matter of CTE patients (Fig. 4D). Thus, immunohistochemical examination revealed characteristics of an axonopathy, including a decreased number of TUBB3-positive and MAG-positive microfilaments. The IHC data were concurrent with Western blot data, indicating that axonal guidance signaling-related proteins are quantitatively reduced in CTE.
After we discovered that the axonal guidance signaling-related proteins are down-regulated in CTE, we further investigated in which cell types these proteins are changed. In this regard, we sorted cell-type specific proteins, ran heat map analyses, and examined alteration of neuronal versus oligodendrocyte-specific proteins in CTE compared to normal control (Fig. 5). We found that neuronal specific proteins such as calmodulin kinase V (CAMKV) and microtubule associated protein 2 (MAP2) (isoform 3) were significantly down-regulated while enolases (ENO1 and 2), well known neuron-specific enzymes, were not changed in CTE (Fig. 5A). On the other hand, most of oligodendrocyte-specific proteins such as 2′,3′-cyclic nucleotide 3′ phosphodiesterase (CNP/CNPase), ectonucleotide pyrophosphatase/phosphodiesterase 6 (ENPP6), ermin (ERMN), and myelin basic protein (MBP) (isoforms 3, 4, and 6) were significantly down-regulated in CTE (Fig. 5B).
In order to find out how 486 up-regulated proteins are associated with the CTE pathogenesis, we further analyzed their molecular interaction/reaction network. The most significant change in KEGG pathways of the up-regulated proteins was the oxidative phosphorylation pathway, an ATP (biological energy) production process in mitochondria (Supplementary Table 5). The oxidative phosphorylation pathway-related 14 proteins included ATP Synthase F1 Subunit Delta (ATP5D), ATP Synthase Membrane Subunit G (ATP5L), ATPase H+ Transporting Accessory Protein 1 (ATP6AP1), ATPase H+ Transporting V0 Subunit C (ATP6V0C), ATPase H+ Transporting V1 Subunit G2 (ATP6V1G2), Mitochondrially Encoded NADH:Ubiquinone Oxidoreductase Core Subunit 5 (MT-ND5), NADH:Ubiquinone Oxidoreductase Subunit AB1 (NDUFAB1), NADH:Ubiquinone Oxidoreductase Subunit A7 (NDUFA7), NADH:Ubiquinone Oxidoreductase Subunit B7 (NDUFB7), NADH:Ubiquinone Oxidoreductase Subunit A12 (NDUFA12), NADH:Ubiquinone Oxidoreductase Subunit A13 (NDUFA13), NADH:Ubiquinone Oxidoreductase Subunit B4 (NDUFB4), NADH:Ubiquinone Oxidoreductase Subunit S6 (NDUFS6), and Succinate Dehydrogenase Complex Flavoprotein Subunit A (SDHA). This data indicates that oxidation of metabolites via the mitochondrial respiratory chain is abnormally modulated in the CTE postmortem brain.
DISCUSSION
CTE is defined by perivascular deposition of hyperphosphorylated protein in the sulcal depths of the cerebral cortex, yet other pathologies, including axonal degeneration, neuroinflammation, microvasculopathy, beta amyloid and TDP-43 deposition are often present [14,17,19,21,26]. Other molecular markers that play active and specific roles in mediating and executing neurodegeneration in CTE have not been fully investigated.
Herein, we performed label-free quantitative proteomic analysis based on protein fractionation by gel electrophoresis coupled to LC-MS/MS of in-gel digested proteins and ion intensity and identified that axonal guidance signaling pathway is significantly affected in the cortex of human brain neuropathologically diagnosed with CTE. The characterization of disease-associated molecules in CTE is complex and involves multiple changes in gene and protein expression in a cell type-specific manner. Indeed, our proteome analysis showed that neuronal and oligodendrocyte-specific protein levels are markedly changed in CTE. The levels of neuronal specific proteins including calmodulin kinase V (CAMKV) and microtubule associated protein 2 (MAP2) (isoform 3) were significantly reduced in CTE. In addition, the levels of major oligodendrocyte-specific proteins including 2′,3′-cyclic nucleotide 3′ phosphodiesterase (CNP/CNPase), myelin basic protein (MBP) (isoforms 3, 4, and 6), ermin (ERMN), and ectonucleotide pyrophosphatase/phosphodiesterase 6 (ENPP6) were significantly reduced in CTE. Interestingly, the protein-protein interaction network analysis showed that cytoskeletal proteins (TUBA1C, TUBA4A, TUBB2A, TUBB3, TUBB4A, TUBB4B, CFL1, and CFL2) are a major cluster of interactome among the axonal guidance signaling pathway in the CTE postmortem brain. In addition, both a cluster of protein kinase- (PRKAR1A, PRKCB, PRKCD, PRKCE, and PRKCG) and a cluster of kinase signaling-related proteins (BCAR1, CDK5, CRK, MAP2K1, and MAP2K2) were markedly associated with impaired axonal guidance signaling pathway in CTE. Otherwise, a protein phosphatase (PPP3CC) was apparently interacted with the cluster of cytoskeletal protein and the cluster of protein kinase- and kinase signaling-related protein, indicating that altered protein phosphatase function may contribute to the dysfunction of axonal signaling pathway regardless of the size of cluster. However, precise interaction mechanisms among these clustered molecules should be further investigated in the future study.
Repetitive head trauma, such as concussive and subconcussive impact injury, produces primary axonal injury through a physical stress and cytoskeletal disruption [37,38,39,40,41,42,43,44]. Axonal injury also triggers molecular signaling pathways that result in axonal degeneration (so called secondary axotomy) [45]. Microtubules are essential to mediate axonal guidance and maintain the structures and long tracts of axons [46,47,48,49,50]. Our current data indicates that TUBB3 (β-tubulin isotype III), a major microtubule exclusively expressed in neurons, is significantly down-regulated in CTE. A growing body of evidence shows that heterozygous missense mutations in
In the present study, we found that oligodendrocyte-derived and myelin-associated proteins such as CNP, myelin-associated glycoprotein (MAG), MBP, and other proteins are significantly down-regulated in moderate to advanced stage of CTE (Stages III and IV), the chronic phase cases. It seems likely that the levels of oligodendrocyte-specific proteins are more affected in CTE than neuronal markers. It is possible that CTE may specifically deregulate protein expression in a cell type-specific manner rather than whole protein expression throughout the brain [54]. Recently, we reported that the OGD-related transcriptome signature is altered in CTE and its alteration is associated with the pathogenesis of CTE [54]. Concurrent with the transcriptome analysis, our proteomic analysis strongly suggests that alteration and deregulation of OGD functions contributes to CTE pathology. OGD function may be involved in the regeneration of axons after mild TBI-induced axonal damage [55]. In contrast, OGD function is severely deregulated in the later stage after repetitive TBI [56]. Interestingly, OGDs possess differential adaptive features in response to the acute or the chronic phases of brain injury [56,57]. OGD dysfunction is characterized by demyelination and white matter pathology. Recent studies indicate that the number of OGDs undergo a dynamic change during the acute phase of fluid percussion brain injury [58]. Because OGD regeneration and axonal remyelination is limited in the injured brain, axonal demyelination leads to consequential long-term sensorimotor deficits and cognitive dysfunction [59].
MAG is a major component of axonal myelination and synthesized by oligodendrocytes. MAG is susceptible to detergent extraction and is associated with the myelin membrane in a sulfatide dependent manner [60]. Regardless of MAG function as a negative regulator of axonal guidance, our data suggest that the level change of MAG protein simply represents the neuropathological marker of CTE at stage III or IV as a failing sign of OGD-dependent regeneration. Our identification of molecules that are linked to the processes of axonal guidance and myelination through neurons and OGDs may provide novel therapeutic targets to restore white matter integrity and ultimately improve neurological symptoms in CTE [61]. Considering that several cell types are affected by trauma and that the pathogenesis of CTE is complex, the crosstalk mechanism between neurons, OGDs, microglia, and astrocytes needs to be further investigated in CTE pathogenesis in future studies.
In conclusion, our proteomic study provides insight on molecules linked to the axonal pathology in CTE. Our findings indicate that the expression of microtubule and myelin ensheathmentrelated proteins is deregulated in both the grey and white matter of CTE. The disruption of axonal integrity and the loss of myelination by oligodendrocytes likely exacerbate axonal damage and demyelination in CTE (Fig. 6). Together, determining mechanisms and therapeutics to maintain the rigid axonal structure by neuronal microtubule assembly and the restoration conditions of myelination by OGD activation will be important strategies in preventing and slowing the neurodegeneration in CTE.
SUPPLEMENTARY INFORMATION
Supplementary Table 1. Information of the postmortem brain tissues included in this study.
Supplementary Table 2. All proteins identified in the cortex of postmortem brains from CTE patients and normal subjects.
Supplementary Table 3. Differentially expressed proteins in CTE brains filtered by fold change cutoff of ≥1.5, p-value cutoff of <0.05 and with minimum of 2 unique peptides for quantification.
Supplementary Table 4. Pathway analysis of differentially expressed proteins in CTE brains.
Supplementary Table 5. KEGG pathway analysis by DAVID (6.8 version) for up-regulated protein signatures in the CTE postmortem brain compared to normal control (p<0.05).
en-28-362-s001.xlsFigures
{kind=link}

{kind=link}

{kind=link}

{kind=link}

{kind=link}
{kind=link}
References
- Thurman DJ, Branche CM, Sniezek JE. The epidemiology of sports-related traumatic brain injuries in the United States: recent developments. J Head Trauma Rehabil 1998;13:1-8.
- Langlois JA, Rutland-Brown W, Wald MM. The epidemiology and impact of traumatic brain injury: a brief overview. J Head Trauma Rehabil 2006;21:375-378.

- Finkelstein E, Corso PS, Miller TR. The incidence and economic burden of injuries in the United States. New York, NY: Oxford University Press, 2006.
- Graves AB, White E, Koepsell TD, Reifler BV, van Belle G, Larson EB, Raskind M. The association between head trauma and Alzheimer's disease. Am J Epidemiol 1990;131:491-501.
- Mortimer JA, French LR, Hutton JT, Schuman LM. Head injury as a risk factor for Alzheimer's disease. Neurology 1985;35:264-267.
- Mortimer JA, van Duijn CM, Chandra V, Fratiglioni L, Graves AB, Heyman A, Jorm AF, Kokmen E, Kondo K, Rocca WA, Shalat SL, Soininen H, EURODEM Risk Factors Research Group. Head trauma as a risk factor for Alzheimer's disease: a collaborative re-analysis of case-control studies. Int J Epidemiol 1991;20:S28-S35.
- O'Meara ES, Kukull WA, Sheppard L, Bowen JD, McCormick WC, Teri L, Pfanschmidt M, Thompson JD, Schellenberg GD, Larson EB. Head injury and risk of Alzheimer's disease by apolipoprotein E genotype. Am J Epidemiol 1997;146:373-384.
- Guo Z, Cupples LA, Kurz A, Auerbach SH, Volicer L, Chui H, Green RC, Sadovnick AD, Duara R, DeCarli C, Johnson K, Go RC, Growdon JH, Haines JL, Kukull WA, Farrer LA. Head injury and the risk of AD in the MIRAGE study. Neurology 2000;54:1316-1323.
- Plassman BL, Havlik RJ, Steffens DC, Helms MJ, Newman TN, Drosdick D, Phillips C, Gau BA, Welsh-Bohmer KA, Burke JR, Guralnik JM, Breitner JC. Documented head injury in early adulthood and risk of Alzheimer's disease and other dementias. Neurology 2000;55:1158-1166.
- Fleminger S, Oliver DL, Lovestone S, Rabe-Hesketh S, Giora A. Head injury as a risk factor for Alzheimer's disease: the evidence 10 years on; a partial replication. J Neurol Neurosurg Psychiatry 2003;74:857-862.
- Omalu BI, DeKosky ST, Minster RL, Kamboh MI, Hamilton RL, Wecht CH. Chronic traumatic encephalopathy in a National Football League player. Neurosurgery 2005;57:128-134.
- Omalu BI, DeKosky ST, Hamilton RL, Minster RL, Kamboh MI, Shakir AM, Wecht CH. Chronic traumatic encephalopathy in a national football league player: part II. Neurosurgery 2006;59:1086-1092.
- McKee AC, Cantu RC, Nowinski CJ, Hedley-Whyte ET, Gavett BE, Budson AE, Santini VE, Lee HS, Kubilus CA, Stern RA. Chronic traumatic encephalopathy in athletes: progressive tauopathy after repetitive head injury. J Neuropathol Exp Neurol 2009;68:709-735.
- McKee AC, Stein TD, Nowinski CJ, Stern RA, Daneshvar DH, Alvarez VE, Lee HS, Hall G, Wojtowicz SM, Baugh CM, Riley DO, Kubilus CA, Cormier KA, Jacobs MA, Martin BR, Abraham CR, Ikezu T, Reichard RR, Wolozin BL, Budson AE, Goldstein LE, Kowall NW, Cantu RC. The spectrum of disease in chronic traumatic encephalopathy. Brain 2013;136:43-64.
- Mez J, Daneshvar DH, Kiernan PT, Abdolmohammadi B, Alvarez VE, Huber BR, Alosco ML, Solomon TM, Nowinski CJ, McHale L, Cormier KA, Kubilus CA, Martin BM, Murphy L, Baugh CM, Montenigro PH, Chaisson CE, Tripodis Y, Kowall NW, Weuve J, McClean MD, Cantu RC, Goldstein LE, Katz DI, Stern RA, Stein TD, McKee AC. Clinicopathological evaluation of chronic traumatic encephalopathy in players of American football. JAMA 2017;318:360-370.
- Omalu B. Chronic traumatic encephalopathy. Prog Neurol Surg 2014;28:38-49.
- McKee AC, Cairns NJ, Dickson DW, Folkerth RD, Keene CD, Litvan I, Perl DP, Stein TD, Vonsattel JP, Stewart W, Tripodis Y, Crary JF, Bieniek KF, Dams-O'Connor K, Alvarez VE, Gordon WA, TBI/CTE group. The first NINDS/NIBIB consensus meeting to define neuropathological criteria for the diagnosis of chronic traumatic encephalopathy. Acta Neuropathol 2016;131:75-86.
- Goldstein LE, Fisher AM, Tagge CA, Zhang XL, Velisek L, Sullivan JA, Upreti C, Kracht JM, Ericsson M, Wojnarowicz MW, Goletiani CJ, Maglakelidze GM, Casey N, Moncaster JA, Minaeva O, Moir RD, Nowinski CJ, Stern RA, Cantu RC, Geiling J, Blusztajn JK, Wolozin BL, Ikezu T, Stein TD, Budson AE, Kowall NW, Chargin D, Sharon A, Saman S, Hall GF, Moss WC, Cleveland RO, Tanzi RE, Stanton PK, McKee AC. Chronic traumatic encephalopathy in blast-exposed military veterans and a blast neurotrauma mouse model. Sci Transl Med 2012;4:134ra60.
- Cherry JD, Tripodis Y, Alvarez VE, Huber B, Kiernan PT, Daneshvar DH, Mez J, Montenigro PH, Solomon TM, Alosco ML, Stern RA, McKee AC, Stein TD. Microglial neuroinflammation contributes to tau accumulation in chronic traumatic encephalopathy. Acta Neuropathol Commun 2016;4:112.
- Cherry JD, Stein TD, Tripodis Y, Alvarez VE, Huber BR, Au R, Kiernan PT, Daneshvar DH, Mez J, Solomon TM, Alosco ML, McKee AC. CCL11 is increased in the CNS in chronic traumatic encephalopathy but not in Alzheimer's disease. PLoS One 2017;12:e0185541.
- McKee AC, Gavett BE, Stern RA, Nowinski CJ, Cantu RC, Kowall NW, Perl DP, Hedley-Whyte ET, Price B, Sullivan C, Morin P, Lee HS, Kubilus CA, Daneshvar DH, Wulff M, Budson AE. TDP-43 proteinopathy and motor neuron disease in chronic traumatic encephalopathy. J Neuropathol Exp Neurol 2010;69:918-929.
- Stern RA, Daneshvar DH, Baugh CM, Seichepine DR, Montenigro PH, Riley DO, Fritts NG, Stamm JM, Robbins CA, McHale L, Simkin I, Stein TD, Alvarez VE, Goldstein LE, Budson AE, Kowall NW, Nowinski CJ, Cantu RC, McKee AC. Clinical presentation of chronic traumatic encephalopathy. Neurology 2013;81:1122-1129.
- Tagge CA, Fisher AM, Minaeva OV, Gaudreau-Balderrama A, Moncaster JA, Zhang XL, Wojnarowicz MW, Casey N, Lu H, Kokiko-Cochran ON, Saman S, Ericsson M, Onos KD, Veksler R, Senatorov VV, Kondo A, Zhou XZ, Miry O, Vose LR, Gopaul KR, Upreti C, Nowinski CJ, Cantu RC, Alvarez VE, Hildebrandt AM, Franz ES, Konrad J, Hamilton JA, Hua N, Tripodis Y, Anderson AT, Howell GR, Kaufer D, Hall GF, Lu KP, Ransohoff RM, Cleveland RO, Kowall NW, Stein TD, Lamb BT, Huber BR, Moss WC, Friedman A, Stanton PK, McKee AC, Goldstein LE. Concussion, microvascular injury, and early tauopathy in young athletes after impact head injury and an impact concussion mouse model. Brain 2018;141:422-458.
- Chen XH, Siman R, Iwata A, Meaney DF, Trojanowski JQ, Smith DH. Long-term accumulation of amyloid-beta, beta-secretase, presenilin-1, and caspase-3 in damaged axons following brain trauma. Am J Pathol 2004;165:357-371.
- Chen XH, Johnson VE, Uryu K, Trojanowski JQ, Smith DH. A lack of amyloid beta plaques despite persistent accumulation of amyloid beta in axons of long-term survivors of traumatic brain injury. Brain Pathol 2009;19:214-223.
- Johnson VE, Stewart W, Smith DH. Traumatic brain injury and amyloid-β pathology: a link to Alzheimer's disease?. Nat Rev Neurosci 2010;11:361-370.
- Marklund N, Blennow K, Zetterberg H, Ronne-Engström E, Enblad P, Hillered L. Monitoring of brain interstitial total tau and beta amyloid proteins by microdialysis in patients with traumatic brain injury. J Neurosurg 2009;110:1227-1237.
- Smith DH, Chen XH, Nonaka M, Trojanowski JQ, Lee VM, Saatman KE, Leoni MJ, Xu BN, Wolf JA, Meaney DF. Accumulation of amyloid beta and tau and the formation of neurofilament inclusions following diffuse brain injury in the pig. J Neuropathol Exp Neurol 1999;58:982-992.
- Smith DH, Chen XH, Iwata A, Graham DI. Amyloid beta accumulation in axons after traumatic brain injury in humans. J Neurosurg 2003;98:1072-1077.
- Stone JR, Okonkwo DO, Singleton RH, Mutlu LK, Helm GA, Povlishock JT. Caspase-3-mediated cleavage of amyloid precursor protein and formation of amyloid beta peptide in traumatic axonal injury. J Neurotrauma 2002;19:601-614.
- Tran HT, LaFerla FM, Holtzman DM, Brody DL. Controlled cortical impact traumatic brain injury in 3xTg-AD mice causes acute intra-axonal amyloid-β accumulation and independently accelerates the development of tau abnormalities. J Neurosci 2011;31:9513-9525.
- Su N, Choi HP, Wang F, Su H, Fei Z, Yang JH, Azadzoi KM. Quantitative proteomic analysis of differentially expressed proteins and downstream signaling pathways in chronic bladder ischemia. J Urol 2016;195:515-523.
- Szklarczyk D, Franceschini A, Kuhn M, Simonovic M, Roth A, Minguez P, Doerks T, Stark M, Muller J, Bork P, Jensen LJ, von Mering C. The STRING database in 2011: functional interaction networks of proteins, globally integrated and scored. Nucleic Acids Res 2011;39:D561-D568.
- Deng W, Wang Y, Liu Z, Cheng H, Xue Y. HemI: a tool-kit for illustrating heatmaps. PLoS One 2014;9:e111988.
- Lee J, Kim CH, Simon DK, Aminova LR, Andreyev AY, Kushnareva YE, Murphy AN, Lonze BE, Kim KS, Ginty DD, Ferrante RJ, Ryu H, Ratan RR. Mitochondrial cyclic AMP response element-binding protein (CREB) mediates mitochondrial gene expression and neuronal survival. J Biol Chem 2005;280:40398-40401.
- Ryu H, Lee J, Impey S, Ratan RR, Ferrante RJ. Anti-oxidants modulate mitochondrial PKA and increase CREB binding to D-loop DNA of the mitochondrial genome in neurons. Proc Natl Acad Sci U S A 2005;102:13915-13920.
- Christman CW, Grady MS, Walker SA, Holloway KL, Povlishock JT. Ultrastructural studies of diffuse axonal injury in humans. J Neurotrauma 1994;11:173-186.
- Johnson VE, Stewart W, Smith DH. Axonal pathology in traumatic brain injury. Exp Neurol 2013;246:35-43.
- Chung RS, Staal JA, McCormack GH, Dickson TC, Cozens MA, Chuckowree JA, Quilty MC, Vickers JC. Mild axonal stretch injury
in vitro induces a progressive series of neurofilament alterations ultimately leading to delayed axotomy. J Neurotrauma 2005;22:1081-1091.
- Jafari SS, Maxwell WL, Neilson M, Graham DI. Axonal cytoskeletal changes after non-disruptive axonal injury. J Neurocytol 1997;26:207-221.
- Maxwell WL, Graham DI. Loss of axonal microtubules and neurofilaments after stretch-injury to guinea pig optic nerve fibers. J Neurotrauma 1997;14:603-614.
- Maxwell WL, Domleo A, McColl G, Jafari SS, Graham DI. Post-acute alterations in the axonal cytoskeleton after traumatic axonal injury. J Neurotrauma 2003;20:151-168.
- Pettus EH, Povlishock JT. Characterization of a distinct set of intra-axonal ultrastructural changes associated with traumatically induced alteration in axolemmal permeability. Brain Res 1996;722:1-11.
- Staal JA, Dickson TC, Gasperini R, Liu Y, Foa L, Vickers JC. Initial calcium release from intracellular stores followed by calcium dysregulation is linked to secondary axotomy following transient axonal stretch injury. J Neurochem 2010;112:1147-1155.
- Greer JE, Hånell A, McGinn MJ, Povlishock JT. Mild traumatic brain injury in the mouse induces axotomy primarily within the axon initial segment. Acta Neuropathol 2013;126:59-74.
- Conde C, Cáceres A. Microtubule assembly, organization and dynamics in axons and dendrites. Nat Rev Neurosci 2009;10:319-332.
- Bradke F, Fawcett JW, Spira ME. Assembly of a new growth cone after axotomy: the precursor to axon regeneration. Nat Rev Neurosci 2012;13:183-193.
- Marner L, Nyengaard JR, Tang Y, Pakkenberg B. Marked loss of myelinated nerve fibers in the human brain with age. J Comp Neurol 2003;462:144-152.
- Coleman M. Axon degeneration mechanisms: commonality amid diversity. Nat Rev Neurosci 2005;6:889-898.
- Baas PW. Microtubule stability in the axon: new answers to an old mystery. Neuron 2013;78:3-5.
- Poirier K, Saillour Y, Bahi-Buisson N, Jaglin XH, Fallet-Bianco C, Nabbout R, Castelnau-Ptakhine L, Roubertie A, Attie-Bitach T, Desguerre I, Genevieve D, Barnerias C, Keren B, Lebrun N, Boddaert N, Encha-Razavi F, Chelly J. Mutations in the neuronal β-tubulin subunit TUBB3 result in malformation of cortical development and neuronal migration defects. Hum Mol Genet 2010;19:4462-4473.
- Demer JL, Clark RA, Tischfield MA, Engle EC. Evidence of an asymmetrical endophenotype in congenital fibrosis of extraocular muscles type 3 resulting from TUBB3 mutations. Invest Ophthalmol Vis Sci 2010;51:4600-4611.
- Tischfield MA, Baris HN, Wu C, Rudolph G, Van Maldergem L, He W, Chan WM, Andrews C, Demer JL, Robertson RL, Mackey DA, Ruddle JB, Bird TD, Gottlob I, Pieh C, Traboulsi EI, Pomeroy SL, Hunter DG, Soul JS, Newlin A, Sabol LJ, Doherty EJ, de Uzcátegui CE, de Uzcátegui N, Collins ML, Sener EC, Wabbels B, Hellebrand H, Meitinger T, de Berardinis T, Magli A, Schiavi C, Pastore-Trossello M, Koc F, Wong AM, Levin AV, Geraghty MT, Descartes M, Flaherty M, Jamieson RV, Møller HU, Meuthen I, Callen DF, Kerwin J, Lindsay S, Meindl A, Gupta ML, Pellman D, Engle EC. Human TUBB3 mutations perturb microtubule dynamics, kinesin interactions, and axon guidance. Cell 2010;140:74-87.
- Seo JS, Lee S, Shin JY, Hwang YJ, Cho H, Yoo SK, Kim Y, Lim S, Kim YK, Hwang EM, Kim SH, Kim CH, Hyeon SJ, Yun JY, Kim J, Kim Y, Alvarez VE, Stein TD, Lee J, Kim DJ, Kim JI, Kowall NW, Ryu H, McKee AC. Transcriptome analyses of chronic traumatic encephalopathy show alterations in protein phosphatase expression associated with tauopathy. Exp Mol Med 2017;49:e333.
- Bradl M, Lassmann H. Oligodendrocytes: biology and pathology. Acta Neuropathol 2010;119:37-53.
- Takase H, Washida K, Hayakawa K, Arai K, Wang X, Lo EH, Lok J. Oligodendrogenesis after traumatic brain injury. Behav Brain Res 2018;340:205-211.
- Flygt J, Djupsjö A, Lenne F, Marklund N. Myelin loss and oligodendrocyte pathology in white matter tracts following traumatic brain injury in the rat. Eur J Neurosci 2013;38:2153-2165.
- Dent KA, Christie KJ, Bye N, Basrai HS, Turbic A, Habgood M, Cate HS, Turnley AM. Oligodendrocyte birth and death following traumatic brain injury in adult mice. PLoS One 2015;10:e0121541.
- Shi H, Hu X, Leak RK, Shi Y, An C, Suenaga J, Chen J, Gao Y. Demyelination as a rational therapeutic target for ischemic or traumatic brain injury. Exp Neurol 2015;272:17-25.
- Pomicter AD, Deloyht JM, Hackett AR, Purdie N, Sato-Bigbee C, Henderson SC, Dupree JL. Nfasc155H and MAG are specifically susceptible to detergent extraction in the absence of the myelin sphingolipid sulfatide. Neurochem Res 2013;38:2490-2502.
- Armstrong RC, Mierzwa AJ, Sullivan GM, Sanchez MA. Myelin and oligodendrocyte lineage cells in white matter pathology and plasticity after traumatic brain injury. Neuropharmacology 2016;110:654-659.