Articles

Article Tools
 View Full Text View Full Text |
 Abstract Abstract |
 Article as PDF Article as PDF |
 Print this Article Print this Article |
 Pubmed Pubmed |
 PMC PMC |
 PubReader PubReader |
 Export to Citation Export to Citation |
 Email Alerts Email Alerts |
 Open Access Open Access |
Share this article on :



Stats or Metrics
Article
Review Article
Exp Neurobiol 2020; 29(3): 177-188
Published online June 30, 2020
https://doi.org/10.5607/en20004
© The Korean Society for Brain and Neural Sciences
Gene Therapy Options as New Treatment for Inherited Peripheral Neuropathy
Rajarathinam Thenmozhi1, Ji-Su Lee2,3, Na Young Park1, Byung-Ok Choi2,3,4* and Young Bin Hong1*
1Department of Biochemistry, College of Medicine, Dong-A University, Busan 49201, 2Stem Cell & Regenerative Medicne Institute, Samsung Medical Center, Seoul 06351, 3Department of Neurology, Samsung Medical Center, Sungkyunkwan University School of Medicine, Seoul 06351, 4Department of Health Sciences and Technology, SAIHST, Sungkyunkwan University, Seoul 06351, Korea
Correspondence to: *To whom correspondence should be addressed.
Young Bin Hong, TEL: 82-51-240-2762, FAX: 82-51-240-2971
e-mail: ybhong@dau.ac.kr
Byung-Ok Choi, TEL: 82-2-3410-1296, FAX: 82-2-3410-0052
e-mail: bochoi@skku.edu
This is an Open Access article distributed under the terms of the Creative Commons Attribution Non-Commercial License (http://creativecommons.org/licenses/by-nc/4.0) which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
Abstract
- Go to
- Abstract
- Graphical Abstract
- INTRODUCTION
- MAJOR TARGETS FOR GENE THERAPY IN IPN
- GENE REPLACEMENT THERAPY
- GENE ADDITION THERAPY
- SUPPRESSION OF MUTANT GENE EXPRESSION
- TARGETING THE GENETIC DOSAGE BY POSTTRANSCRIPTIONAL MODULATION
- MODULATION OF TRANSCRIPTIONAL ACTIVITY
- OPTIMIZATION OF THERAPEUTIC STRATEGY FOR IPN TREATMENT
- CONCLUSION AND PERSPECTIVES
- ACKNOWLEDGEMENTS
- CONFLICT OF INTEREST
- Figure
- Table
- Reference
Inherited peripheral neuropathy (IPN) is caused by heterogeneous genetic mutations in more than 100 genes. So far, several treatment options for IPN have been developed and clinically evaluated using small molecules. However, gene therapy-based therapeutic strategies have not been aggressively investigated, likely due to the complexities of inheritance in IPN. Indeed, because the majority of the causative mutations of IPN lead to gainof- function rather than loss-of-function, developing a therapeutic strategy is more difficult, especially considering gene therapy for genetic diseases began with the simple idea of replacing a defective gene with a functional copy. Recent advances in gene manipulation technology have brought novel approaches to gene therapy and its clinical application for IPN treatment. For example, in addition to the classically used gene replacement for mutant genes in recessively inherited IPN, other techniques including gene addition to modify the disease phenotype, modulations of target gene expression, and techniques to edit mutant genes have been developed and evaluated as potent therapeutic strategies for dominantly inherited IPN. In this review, the current status of gene therapy for IPN and future perspectives will be discussed.
Graphical Abstract
- Go to
- Abstract
- Graphical Abstract
- INTRODUCTION
- MAJOR TARGETS FOR GENE THERAPY IN IPN
- GENE REPLACEMENT THERAPY
- GENE ADDITION THERAPY
- SUPPRESSION OF MUTANT GENE EXPRESSION
- TARGETING THE GENETIC DOSAGE BY POSTTRANSCRIPTIONAL MODULATION
- MODULATION OF TRANSCRIPTIONAL ACTIVITY
- OPTIMIZATION OF THERAPEUTIC STRATEGY FOR IPN TREATMENT
- CONCLUSION AND PERSPECTIVES
- ACKNOWLEDGEMENTS
- CONFLICT OF INTEREST
- Figure
- Table
- Reference
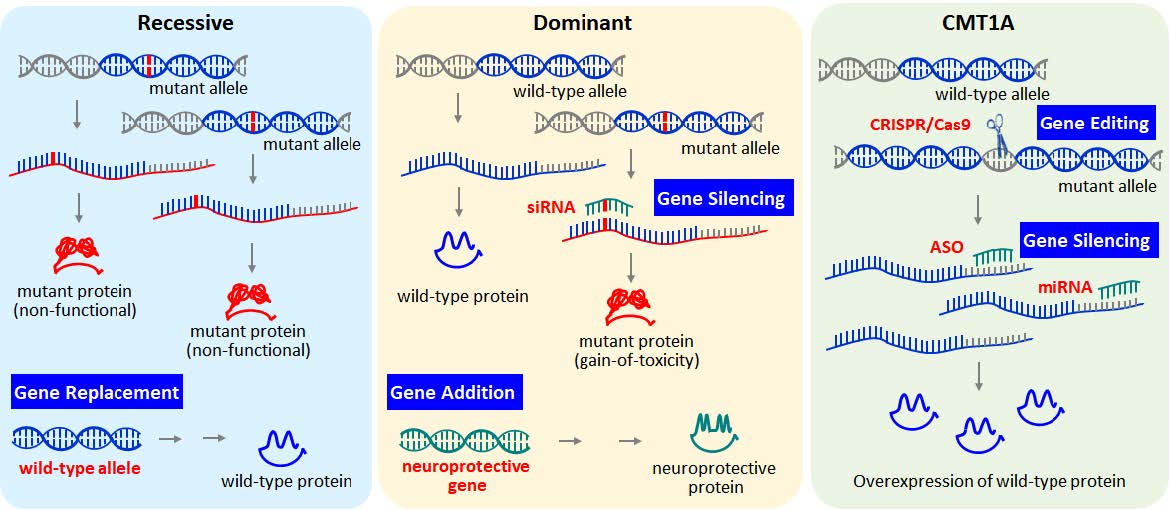
Keywords: Inherited peripheral neuropathy, Gene therapy, Antisense oligonucleotide, miRNA, Gene editing
INTRODUCTION
- Go to
- Abstract
- Graphical Abstract
- INTRODUCTION
- MAJOR TARGETS FOR GENE THERAPY IN IPN
- GENE REPLACEMENT THERAPY
- GENE ADDITION THERAPY
- SUPPRESSION OF MUTANT GENE EXPRESSION
- TARGETING THE GENETIC DOSAGE BY POSTTRANSCRIPTIONAL MODULATION
- MODULATION OF TRANSCRIPTIONAL ACTIVITY
- OPTIMIZATION OF THERAPEUTIC STRATEGY FOR IPN TREATMENT
- CONCLUSION AND PERSPECTIVES
- ACKNOWLEDGEMENTS
- CONFLICT OF INTEREST
- Figure
- Table
- Reference
Inherited peripheral neuropathy (IPN) is caused by genetic mutations which damage the integrity of either the axon or myelin of peripheral nerves. The prominent clinical phenotypes of IPN include progressive and symmetrical distal weakness resulting in loss of sensation, muscle wasting and gait disturbances. The symptom appears in a length dependent manner that muscle weakness and atrophy was observed from distal to proximal parts of all limbs [1]. The defects in the peripheral nerves are more severe in the distal part than in the proximal part because smaller diameter of the nerves renders more vulnerable to the degeneration. Clinically, IPN can be subdivided into hereditary motor and sensory neuropathy or Charcot-Marie-Tooth disease, distal hereditary motor neuropathy, and hereditary sensory and autonomic neuropathy [2]. The total number of IPN patients is estimated to be more than three million globally.
Currently, the treatment options for IPN are very limited. Although several attempts have been made to lessen or ameliorate the disease phenotype after validating the efficacy of animal studies, clinical benefits remain uncertain. For example, vitamin C was proven to be successful in rodent models, however, its efficacy could not be duplicated in clinical trials [3,-5]. Recently, PXT3003, a novel combination of baclofen, naltrexone hydrochloride and D-sorbitol is under clinical evaluation, yet the clinical benefits need further elucidation [6,-8]. Indeed, unsatisfactory outcomes in clinical practice could be attributed to inappropriate treatments. Up to now, treatment approaches have focused on modulating the disease phenotype by indirectly reducing the expression of toxic protein and/or enhancing myelination and axonal integrity. Therefore, the possibility of direct manipulation of mutant gene expression should be considered as an acceptable and effective therapeutic option.
Although IPN was first described in the 19th century, the first causative gene was isolated in 1991 [9,-12]. By the advent of next-generation sequencing technology, however, more than 100 distinct genes have been identified as causative genes for IPN [13, 14]. Among the numerous causative genes, peripheral myelin protein 22 (PMP22), myelin protein zero (MPZ), gap junction protein beta 1 (GJB1) and mitofusin2 (MFN2) are prevalent in over 80% of genetically isolated IPN patients [15,-18]. As such, the majority of research has been focused on these genes, revealing the pathophysiological mechanism, and developing therapeutic options.
Gene therapy is a fundamental and straightforward strategy to overcome the genetic defects in inherited disorders. Indeed, replacing a mutant gene with a functional copy through gene delivery might be the ultimate treatment strategy for IPN. Everyday gene therapy treatment options are expanding, by virtue of novel technical advances in gene manipulation. Current strategies for gene therapy can be categorized into four types: gene replacement, gene addition, gene knockdown or modulation of gene expression, and gene editing or correction [19,-22]. Besides the simple delivery of functional genes, the ability to manipulate the expression of mutant genes with toxic gain-of-function, or to correct mutant genes into functional genes, is now possible. Although gene therapy has not been clinically investigated in IPN patients, a breakthrough in gene therapy with benefits for IPN is anticipated.
MAJOR TARGETS FOR GENE THERAPY IN IPN
- Go to
- Abstract
- Graphical Abstract
- INTRODUCTION
- MAJOR TARGETS FOR GENE THERAPY IN IPN
- GENE REPLACEMENT THERAPY
- GENE ADDITION THERAPY
- SUPPRESSION OF MUTANT GENE EXPRESSION
- TARGETING THE GENETIC DOSAGE BY POSTTRANSCRIPTIONAL MODULATION
- MODULATION OF TRANSCRIPTIONAL ACTIVITY
- OPTIMIZATION OF THERAPEUTIC STRATEGY FOR IPN TREATMENT
- CONCLUSION AND PERSPECTIVES
- ACKNOWLEDGEMENTS
- CONFLICT OF INTEREST
- Figure
- Table
- Reference
Phenotypically, IPN can be divided into three types according to the origin of degeneration: axonal, demyelinating, and intermediate type. Axonal type is primarily caused by degeneration of axon and demyelinating type is caused by malfunction of myelinating Schwann cell. Intermediate type shows both axonal and demyelinating features (Table 1). Thus major targets for IPN treatment has focused on either enhancing myelination of Schwann cell for demyelinating type or increasing the mitochondrial activity and axonal transport for axonal type. To enhance the myelination of Schwann cell, ascorbic acid, curcumin, a HSP90 inhibitor, and a progesterone antagonist have been evaluated in preclinical or clinical studies [3, 23,-25]. Recently, a combination of preexisting drugs, PXT3003, was developed by the application of systems biology and its efficacy is currently under clinical evaluation after demonstration of the myelination enhancement in animal model [6,-8]. In contrast, improvement of mitochondrial metabolism or axonal transport have been paid attention to manage the axonal type of IPN. Coenzyme Q10 and a mitofusin agonist, which might be associated the mitochondrial activity, ameliorated the axonopathic phenotype in MFN2-mutated IPN [26, 27]. In addition, inhibition of histone deacetylase 6 (HDAC6) increased acetylated alpha-tubulin and improved axonal transport in HSPB1 or GARS animal models [28,-31].
As more than 100 genes have been isolated as causative of IPN, clinical interest has not been able to investigate every gene. Instead, mutations in several genes (PMP22, MPZ, and MFN2) have received major attention for therapeutic applications [4, 24,32,-34]. Interestingly, the mutations in those genes are all dominantly inherited. Indeed, more than 95% of CMT cases are dominantly inherited, with the remaining 5% of cases inherited via the autosomal recessive or X-linked recessive manner. As such, classical gene therapy, a gene replacement, cannot be applied to most IPN patients.
Recent advances in gene manipulation technology shed light on the development of novel gene therapy strategies for dominantly inherited IPN. For example, mutant alleles can be selectively suppressed while normal alleles can regenerate the damaged peripheral nerve. Duplication of PMP22, is the major cause of demyelinating type IPN, and comprises over 40% of all IPN cases. In this subtype, the duplication of PMP22 in one haploid causes the total expression of PMP22 protein to be 1.5 times higher than normal, which in turn causes cellular stress and the demyelination of Schwann cells [35]. Thus, reducing the expression level of PMP22 protein in patients has been the major target of IPN treatment. Reduction of gene expression can be achieved by impairing the transcription and degrading the mRNA transcript, or by blocking the protein translation. Recently, gene editing technology has also proposed the conversion of a mutated gene into a normal copy might be the ultimate therapeutic option for genetic diseases (Fig. 1).
GENE REPLACEMENT THERAPY
- Go to
- Abstract
- Graphical Abstract
- INTRODUCTION
- MAJOR TARGETS FOR GENE THERAPY IN IPN
- GENE REPLACEMENT THERAPY
- GENE ADDITION THERAPY
- SUPPRESSION OF MUTANT GENE EXPRESSION
- TARGETING THE GENETIC DOSAGE BY POSTTRANSCRIPTIONAL MODULATION
- MODULATION OF TRANSCRIPTIONAL ACTIVITY
- OPTIMIZATION OF THERAPEUTIC STRATEGY FOR IPN TREATMENT
- CONCLUSION AND PERSPECTIVES
- ACKNOWLEDGEMENTS
- CONFLICT OF INTEREST
- Figure
- Table
- Reference
As most genetic diseases are caused by a single gene defect, replacement of the defective gene is a straightforward approach, and the majority of gene therapy research focuses on gene replacement for recessive genetic disorders. Recently, several gene replacement methods for autosomal recessive and X-linked cases of IPN have been investigated by a European research group.
For autosomal recessive cases of IPN, Schiza et al. [36] evaluated the efficacy of gene replacement therapy for the SH3TC2 (SH3 domain and tetratricopeptide repeats 2) mutation in IPN. SH3TC2 protein is predominantly expressed in myelinating Schwann cells, and the loss-of-function mutations in the SH3TC2 gene contribute to onset of CMT type 4C recessively inherited demyelinating neuropathy [37]. Schiza et al. [36] generated a lentiviral vector expressing the SH3TC2 gene under the control of an MPZ promoter, a Schwann cell specific promoter. The engineered lentivirus expressing the target gene was effectively delivered to Schwann cells via intrathecal injection in a Sh3tc2-/- mouse model and rescued the neuropathic phenotype. After 8 weeks, the mutant mice, exhibited improved myelination in the lumbar spinal roots and sciatic nerves and the motor behavior was also enhanced.
Intriguingly, the same group also tried gene replacement therapy in X-linked dominant type IPN. There, GJB1 gene mutations cause loss of Connexin 32 (Cx32) in gap junctions and lead to a severe form of inherited demyelinating CMTX1 neuropathy [38]. The mutations in GJB1 cause dysfunction in the Cx32 protein localized in the paranodal loops of non-compact myelin and the Schmidt–Lanterman incisures of Schwann cells [39]. Although the GJB1 mutation caused phenotype is considered to be dominantly inherited, the clinical phenotypes are dramatically different according to gender. An affected female with a heterozygous GJB1 mutation exhibits later onset and milder phenotype than an affected male with hemizygosity due to X-inactivation [40]. To validate the therapeutic effect of gene replacement, they utilized GJB1-null/Cx32 knockout (KO) mice, which exhibit severe demyelination, as well as inflammation in the peripheral nerve [41,-43]. Intraneural injection of lentivirus expressing GJB1 by MPZ promoter (LV. Mpz-GJB1) before the phenotype onset in GJB1-null mice significantly reduced the inflammation and ameliorated the peripheral neuropathic phenotype [41]. In a follow-up study, Kagiava et al. [42] also validated the efficacy of intrathecal delivery. Intrathecal administration is less invasive than intraneural delivery, and as a result the clinical feasibility of gene therapy is improved. Recently, Kagiava et al. [43] also demonstrated a therapeutic benefit even if gene therapy is performed after the onset of peripheral neuropathic symptoms. Collectively, these results increase the possibility of success in future clinical trials and positive outcomes for CMT1X patients.
GENE ADDITION THERAPY
- Go to
- Abstract
- Graphical Abstract
- INTRODUCTION
- MAJOR TARGETS FOR GENE THERAPY IN IPN
- GENE REPLACEMENT THERAPY
- GENE ADDITION THERAPY
- SUPPRESSION OF MUTANT GENE EXPRESSION
- TARGETING THE GENETIC DOSAGE BY POSTTRANSCRIPTIONAL MODULATION
- MODULATION OF TRANSCRIPTIONAL ACTIVITY
- OPTIMIZATION OF THERAPEUTIC STRATEGY FOR IPN TREATMENT
- CONCLUSION AND PERSPECTIVES
- ACKNOWLEDGEMENTS
- CONFLICT OF INTEREST
- Figure
- Table
- Reference
Although demyelinating neuropathy occurs mainly due to dominant inheritance, one research group has consistently developed a phenotype modulating strategy using a neurotrophic factor. Neurotrophic factors such as nerve growth factor, brain-derived neurotrophic factor (BDNF), neurotrophin-3 (NT-3) and neurotrophin-4/5 are well-known to bind to protein tyrosine kinase receptor and activate the downstream signaling pathways in neuronal cells [44]. In addition, these neurotrophic factors also influence the survival and differentiation of Schwann cell [45]. The formation of peripheral nerve is a complex and dynamic process between neuronal axon and Schwann cell. The interaction between these cells leads to the proliferation, migration, and myelination of Schwann cell as well as axon development. During the process neurotrophic factors such as BDNF and NT-3 play an important role in the myelination and axonal growth [46, 47].
When NT-3 was subcutaneously administered into a Tr-J mouse model, a mouse model of demyelinating neuropathy with naturally occurring Leu16Pro mutation in the PMP22 gene, elevated numbers of myelinated fiber forming regeneration units were observed along with axonal regeneration [48]. In the same report, the clinical efficacy of NT-3 in CMT1A patients was evaluated. The patients treated with NT-3 exhibited enhanced nerve regeneration of the sural nerve. In a follow-up study, the administration of agonistic antibodies to NT-3 receptors TrkB and TrkC improved the neuropathic phenotype of Tr-J mice [49]. In addition, because long-term treatment with NT-3 is not clinically achievable due to its short half-life, gene delivery of NT-3 by recombinant adeno-associated virus (rAAV) was investigated [50]. Indeed, intramuscular delivery of rAAV-NT-3 sustained the release of NT-3, as well as promoted active myelination and nerve regeneration in Tr-J mice. The clinical benefit of neurotrophic factors in modulating the disease pathogenesis of demyelinating neuropathy was also observed by other researchers. For example, the administration of neuregulin-1 enhanced myelination by stimulating myelination pathways in rodent models [51, 52].
SUPPRESSION OF MUTANT GENE EXPRESSION
- Go to
- Abstract
- Graphical Abstract
- INTRODUCTION
- MAJOR TARGETS FOR GENE THERAPY IN IPN
- GENE REPLACEMENT THERAPY
- GENE ADDITION THERAPY
- SUPPRESSION OF MUTANT GENE EXPRESSION
- TARGETING THE GENETIC DOSAGE BY POSTTRANSCRIPTIONAL MODULATION
- MODULATION OF TRANSCRIPTIONAL ACTIVITY
- OPTIMIZATION OF THERAPEUTIC STRATEGY FOR IPN TREATMENT
- CONCLUSION AND PERSPECTIVES
- ACKNOWLEDGEMENTS
- CONFLICT OF INTEREST
- Figure
- Table
- Reference
As a mutated gene is translated into mutant proteins via an mRNA intermediate, inhibiting the translation of mutant mRNA into mutant protein can also be a potential therapeutic target for IPN. For this strategy, utilization of RNA interference (RNAi) has been well studied. Small interfering RNA (siRNA) is a short double-stranded RNA approximately 19~22 nucleotides long [53] which can nullify gene expression by breaking down the mRNA transcripts in a sequence-specific manner. Recently, siRNA-based technique has been a powerful research tool for gene silencing in both basic and therapeutic research [54, 55]. By introducing siRNA or short hairpin RNA (shRNA), gene expression level can be successfully modulated. In addition, because of sequence-specific mRNA breakdown, discrimination between mutant alleles and wild-type sequence is simply achieved. Since dominantly inherited genetic disorders are caused by toxic gain-of-function mutations rather than loss-of-function mutations in recessively inherited genetic disorders, mutant allele-specific targeting ought to be the primary strategy, rather than the addition of normal genes. Indeed, siRNAs have been proven to be successful at the specific targeting and silencing of the mutant allele in dominantly inherited disorders including neurodegenerative diseases such as Alzheimer’s disease, Parkinson’s disease, Huntington’s disease, Machado-Joseph disease, and amyotrophic lateral sclerosis [56,-60].
For IPN treatment, Lee et al. [61] evaluated the efficacy of mutant allele-specific siRNA using Tr-J mice. Lee et al. [61] designed and isolated the mutant allele (c.47T>C, p.Leu16Pro in mouse Pmp22)-specific siRNA for Tr-J mice and evaluated the potency of allele specificity, both
TARGETING THE GENETIC DOSAGE BY POSTTRANSCRIPTIONAL MODULATION
- Go to
- Abstract
- Graphical Abstract
- INTRODUCTION
- MAJOR TARGETS FOR GENE THERAPY IN IPN
- GENE REPLACEMENT THERAPY
- GENE ADDITION THERAPY
- SUPPRESSION OF MUTANT GENE EXPRESSION
- TARGETING THE GENETIC DOSAGE BY POSTTRANSCRIPTIONAL MODULATION
- MODULATION OF TRANSCRIPTIONAL ACTIVITY
- OPTIMIZATION OF THERAPEUTIC STRATEGY FOR IPN TREATMENT
- CONCLUSION AND PERSPECTIVES
- ACKNOWLEDGEMENTS
- CONFLICT OF INTEREST
- Figure
- Table
- Reference
CMT1A is the most common type of IPN resulting in the demyelination of Schwann cells due to a 1.5-fold overexpression of PMP22 myelinating protein. Because over 40% of all IPN cases have CMT1A, most research has focused on exploring the novel agents capable of decreasing the expression level of PMP22. By developing a high-throughput screening method and with the aid of systems biology, some research groups were able to isolate or repurpose several drugs or drug combinations useful for downregulating PMP22 expression [6,-8, 62]. However, the mode-of-action as well as the potency for these drugs in downregulating PMP22 expression remains unclear. Alternatively, two independent groups have developed novel gene therapies which directly manipulate the gene dosage of PMP22. One group isolated novel microRNAs (miRNAs) which specifically target the 3'-UTR of PMP22 mRNA and the other screened antisense oligonucleotides (ASO) which successfully downregulate PMP22 levels.
MicroRNAs (miRNA) are endogenous small noncoding RNAs, approximately 22 nucleotides in length [63], which readily bind to the 3'-UTR of target mRNAs and induce degradation. Thus miRNAs can regulate gene expression by acting as modifiers to silence overexpressed genes. The significance of regulatory function of miRNAs in the development of the peripheral nervous system has been investigated. Ablation or reduction of Dicer from Schwann cells can impair normal myelination and axonal integrity [64,-67].
Regarding PMP22 gene expression, several miRNAs such as miR-9 and miR-29b are known to post-transcriptionally target the 3'-UTR of PMP22 [68]. Since miRNAs have great potential in regulating the expression level of target mRNAs, targeting PMP22 with its specific miRNA might be an excellent therapeutic option for controlling CMT1A caused by PMP22 overexpression. In this context, Lee et al. [69] reported that the administration of miRNAs downregulated the PMP22 expression levels in a CMT1A mouse model. Indeed, the expression level of several miRNAs were changed and miR-381 and miR-9 can modulate the expression level of PMP22. Using the lentiviral system, LV-miR-381 as well as LV-miR-9 were administered into the sciatic nerve of a C22 mouse, which harbors 7 copies of the human PMP22 gene and an expression level of hPMP22 1.7 fold higher than mouse Pmp22 [70,-72]. Expression of both miR-381 and miR-9 enhance the locomotor function, electrophysiological integrity (motor nerve conduction velocity and compound action potential), and myelination through the reduction of PMP22 levels in the sciatic nerve of C22 mice. This report revealed a new way for developing potential IPN therapeutic strategies by using miRNA-mediated regulation of gene expression.
RNA transcripts can also be modulated by ASOs; which are synthetic nucleic acids in a single strand capable of binding to the target mRNA resulting in degradation, interference with pre-mRNA processing or protein binding, and alteration of RNA structure [73]. Recently, the application of ASOs has become an emerging tool to manage various degenerative neuromuscular diseases. The clinical application of ASOs has exhibited successful outcomes in spinal muscular atrophy (SMA) and Duchenne’s muscular dystrophy (DMD) by modulating the splicing of the mRNA [74,-78]. Developments of ASO therapy for these diseases have shifted the strategic paradigms of gene therapy. SMA is caused by mutations in survival motor neuron 1 (SMN1) gene with autosomal recessive inheritance pattern. In humans, an evolutionarily duplicated gene, SMN2, possesses almost identical nucleotide sequence to SMN1. However, a critical substitution at position 6 (C to T) of exon 7 in SMN2 causes aberrant splicing and degradation of mRNA [79]. Instead of correcting the mutated SMN1 in the patients, ASO therapy targets to recuperate SMN2 function by intervening the aberrant splicing. After identification of intron splicing silencer N1 (ISS-N1) sequence in intron 7 that is critical in the skipping of exon 7 in SMN2, ISS-N1 sequence-inhibiting ASO has been developed and successfully demonstrated the therapeutic efficacy through clinical studies [75, 76]. On the other hand, DMD gene is one of the largest human genes with 79 exons and 14kb transcripts. Generally, out-of-frame mutations in DMD gene result in DMD, and in-frame mutations lead to Becker muscular dystrophy (BMD) exhibiting a milder phenotype compared to DMD. Although DMD is an X-linked recessive disease, the huge length of the causative gene renders it difficult to treat with simple gene replacement therapy. Instead, exon skipping strategy using ASO targets and induces the deletion of exon 51 of DMD mRNA during splicing, which then results in the production of shortened but functional dystrophin protein and conversion of out-of-frame mutation into in-frame mutation. Accordingly, the DMD phenotype can be ameliorated as BND phenotype by treatment of ASO [77, 78].
The suppression of PMP22 expression can be achieved by hybridizing the ASO to result in the specific inhibition and degradation of PMP22 through endogenous RNase H activity. Zhao et al. [80] investigated the potency of PMP22 targeting ASOs using two rodent models for CMT1A. After ASO treatment, both mouse and rat models of CMT1A showed a 35% reduction in PMP22 mRNA, which reduced disease progression and improved CMT1 phenotypes. Zhao et al. [80] also proposes that skin biopsy samples are ideal for detecting the mRNA level of PMP22 as a useful biomarker for future clinical trials on CMT1A.
MODULATION OF TRANSCRIPTIONAL ACTIVITY
- Go to
- Abstract
- Graphical Abstract
- INTRODUCTION
- MAJOR TARGETS FOR GENE THERAPY IN IPN
- GENE REPLACEMENT THERAPY
- GENE ADDITION THERAPY
- SUPPRESSION OF MUTANT GENE EXPRESSION
- TARGETING THE GENETIC DOSAGE BY POSTTRANSCRIPTIONAL MODULATION
- MODULATION OF TRANSCRIPTIONAL ACTIVITY
- OPTIMIZATION OF THERAPEUTIC STRATEGY FOR IPN TREATMENT
- CONCLUSION AND PERSPECTIVES
- ACKNOWLEDGEMENTS
- CONFLICT OF INTEREST
- Figure
- Table
- Reference
Reducing the protein expression of PMP22 can also be achieved by modulating the transcriptional activity of a gene. Recently, two independent research groups demonstrated the feasibility of PMP22 reduction by disrupting either the promoter or enhancer of PMP22 with gene editing technology [81, 82]. The clustered regularly interspaced short palindromic repeats (CRISPR) and related Cas genes are now emerging as essential tools for gene editing [83, 84]. Briefly, DNA from viruses or plasmids are cut into small fragments and integrated into a CRISPR locus with a series of short repeats, around 20 bps. The loci are transcribed, and later the transcripts are processed to generate small RNAs to target foreign DNA based on the sequence complementarity principle. Using this groundbreaking new technology, numerous clinical applications have been attempted to treat various types of diseases.
Intriguingly, gene editing technology was applied to disrupt normal genes rather than to correct the mutant gene for CMT1A treatment. Pantera et al. [81] investigated the feasibility of gene editing for reducing the transcription of PMP22
The
OPTIMIZATION OF THERAPEUTIC STRATEGY FOR IPN TREATMENT
- Go to
- Abstract
- Graphical Abstract
- INTRODUCTION
- MAJOR TARGETS FOR GENE THERAPY IN IPN
- GENE REPLACEMENT THERAPY
- GENE ADDITION THERAPY
- SUPPRESSION OF MUTANT GENE EXPRESSION
- TARGETING THE GENETIC DOSAGE BY POSTTRANSCRIPTIONAL MODULATION
- MODULATION OF TRANSCRIPTIONAL ACTIVITY
- OPTIMIZATION OF THERAPEUTIC STRATEGY FOR IPN TREATMENT
- CONCLUSION AND PERSPECTIVES
- ACKNOWLEDGEMENTS
- CONFLICT OF INTEREST
- Figure
- Table
- Reference
In accordance with the recent advances in gene manipulation technology, novel gene therapy was developed and evaluated for IPN using animal models (Table 2). To effectively translate the plausible preclinical results from gene therapy into clinical benefits for IPN patients, several aspects, such as securing the efficacy and safety, should be considered.
Securing safety is a first-line consideration for gene therapy. Although viral delivery provides the feasibility of tissue-specific targeting, long-term effects, and a large capacity for the cargo gene, they still possess the risk of virulence-mediated immunotoxicity and genotoxicity which may impede therapeutic outcomes. Innate and adaptive immune responses to the delivered vectors or the transgene are substantial challenges to the safety of the gene therapy. Although AAV is known to cause relatively weak inflammatory responses compared to other viral vectors, the possibility of activation of T cell or antigen presenting cell by AAV administration still needs to be improved for clinical trials. Genotoxicity from gene delivery includes insertional mutagenesis, disruption of untargeted gene, and activation of proto-oncogenes depending on the virus type, target cells, and target sequences. Recently, several strategies have been devised to prevent the viral vector-mediated genotoxicity [85]. Activation of proto-oncogenes can be reduced by self-inactivation vector and chromatin insulator. Disruption of U3 in 3'-LTR (long terminal repeat) of the lentivirus reduced the promoter and enhancer activity on the neighboring gene [86]. Insertion of insulator after enhancer sequence in the viral vector can also block the activation of transcriptionally silent proto-oncogene [87]. The long-term effect of the delivered gene sometime causes deleterious outcomes due to uncontrolled expression. Thus regulation of the transgene expression using a molecular switch such as tetracycline-controlled transcriptional activation in animal study might be helpful to increase the safety of gene therapy in clinical application. Recently, introduction of a type III hammerhead ribozyme (HHR) at the 3'-UTR of the transgene exhibited the potency in regulating the protein expression. Since HHR possesses cis-cleaving activity, the cleavage of the 3'-UTR by HHR resulted in the degradation of transgene mRNA. Co-application of a ASO targeting HHR sequence inhibited HHR activity thereby allowing the protein expression [88]. Compared to the viral gene therapy, non-viral delivery has safety advantage. However, the cytotoxicity of their vehicle composition should be thoroughly evaluated. In addition, risk assessments for horizontal or vertical transmission should also be followed for clearing the genotoxicity concern in gene therapy.
To improve the efficacy of gene therapy, design of delivery vectors and administration route should be carefully deliberated. Especially, tropism of viral vectors is important in the effective delivery of the transgene into the target cells. Since the main target of peripheral neuropathy are Schwann cells or neuronal axon, selection of viral serotype is properly considered. Recently, the transduction efficiency of AAV and Lentivirus on the Schwann cells was compared [89]. Lentivirus showed the highest transduction efficiency on both rat and human Schwann cells, while AAV showed entirely different efficiency according to serotypes. AAV1 showed the highest transduction efficiency in rat Schwann cells, whereas AAV2 and AAV6 showed better potency on human Schwann cells. According to another previous study using mice, AAV1 transduced both Schwann cells and neurons while AAV2 and AAV8 showed selective preference on sensory neuron and Schwann cells, respectively [90]. In this regard, all the viral vector-mediated gene therapy in IPN utilized either lentivirus or AAV1. Thus further optimization of the vector system might increase the possibility of successful translation of gene therapy from preclinical studies to future clinical trials.
Increasing the target specificity is also significant issue for non-viral gene delivery. To improve transfection efficiency, increasing DNA condensation and stability, incorporating cell penetrating peptide, facilitating endosome escape, and increasing nuclear uptake and translocation by manipulating chemical composition can be considered during the design of non viral gene delivery system [91]. Recently, application of enzymes (e.g. MMP), cell specific antibodies (e.g. anti-HER2 and anti-CD,3 antibody), and aptamers have been developed to increase the target specificity [92,-94]. Thus development of novel specific targets for Schwann cell or peripheral neuron is needed to increase the transfection efficiency of non-viral vectors for IPN gene therapy.
Determining the delivery route is also an important part in the efficacy and feasibility of gene therapy. Because IPN affects the peripheral nerves, the highest efficacy could be achieved via an intraneural delivery. However, the direct administration of therapeutics into a peripheral nerve can cause tissue damage and may worsen the disease phenotype. As such, intrathecal or subcutaneous delivery provide an alternative option for IPN treatment delivery.
CONCLUSION AND PERSPECTIVES
- Go to
- Abstract
- Graphical Abstract
- INTRODUCTION
- MAJOR TARGETS FOR GENE THERAPY IN IPN
- GENE REPLACEMENT THERAPY
- GENE ADDITION THERAPY
- SUPPRESSION OF MUTANT GENE EXPRESSION
- TARGETING THE GENETIC DOSAGE BY POSTTRANSCRIPTIONAL MODULATION
- MODULATION OF TRANSCRIPTIONAL ACTIVITY
- OPTIMIZATION OF THERAPEUTIC STRATEGY FOR IPN TREATMENT
- CONCLUSION AND PERSPECTIVES
- ACKNOWLEDGEMENTS
- CONFLICT OF INTEREST
- Figure
- Table
- Reference
As the incidence of CMT1A is highest in IPN with therapeutic benefits over millions of patients, therapeutic strategies for suppressing PMP22 expression urgently need to be proven safe for patients. Gene suppression mediated therapeutic strategies require relatively short nucleotides compared to the delivery of a whole gene, which enables
Novel therapeutic options for IPN have been developed by virtue of the breakthroughs in RNA interference, oligonucleotide-based therapy, and genome editing technology. The development of a novel therapeutic option for CMT1A could be beneficial to the many patients affected by PMP22. Although it may be a long way until this seemingly straightforward concept comes into reality, these meaningful innovations are expected to greatly broaden the scope of gene therapy in the near future.
ACKNOWLEDGEMENTS
- Go to
- Abstract
- Graphical Abstract
- INTRODUCTION
- MAJOR TARGETS FOR GENE THERAPY IN IPN
- GENE REPLACEMENT THERAPY
- GENE ADDITION THERAPY
- SUPPRESSION OF MUTANT GENE EXPRESSION
- TARGETING THE GENETIC DOSAGE BY POSTTRANSCRIPTIONAL MODULATION
- MODULATION OF TRANSCRIPTIONAL ACTIVITY
- OPTIMIZATION OF THERAPEUTIC STRATEGY FOR IPN TREATMENT
- CONCLUSION AND PERSPECTIVES
- ACKNOWLEDGEMENTS
- CONFLICT OF INTEREST
- Figure
- Table
- Reference
This study was supported by the National Research Foundation of Korea (NRF) grants funded by the Korean government, MSIP (NRF-2016R1A5A2007009, NRF-2018R1A4A1024506 and NRF-2019R1F1A1060313), and by the Korean Health Technology R&D Project, Ministry of Health & Welfare (HI14C3484 and HI16C0426).
CONFLICT OF INTEREST
- Go to
- Abstract
- Graphical Abstract
- INTRODUCTION
- MAJOR TARGETS FOR GENE THERAPY IN IPN
- GENE REPLACEMENT THERAPY
- GENE ADDITION THERAPY
- SUPPRESSION OF MUTANT GENE EXPRESSION
- TARGETING THE GENETIC DOSAGE BY POSTTRANSCRIPTIONAL MODULATION
- MODULATION OF TRANSCRIPTIONAL ACTIVITY
- OPTIMIZATION OF THERAPEUTIC STRATEGY FOR IPN TREATMENT
- CONCLUSION AND PERSPECTIVES
- ACKNOWLEDGEMENTS
- CONFLICT OF INTEREST
- Figure
- Table
- Reference
The authors report no conflict of interest with any person or Institute.
Figures
- Go to
- Abstract
- Graphical Abstract
- INTRODUCTION
- MAJOR TARGETS FOR GENE THERAPY IN IPN
- GENE REPLACEMENT THERAPY
- GENE ADDITION THERAPY
- SUPPRESSION OF MUTANT GENE EXPRESSION
- TARGETING THE GENETIC DOSAGE BY POSTTRANSCRIPTIONAL MODULATION
- MODULATION OF TRANSCRIPTIONAL ACTIVITY
- OPTIMIZATION OF THERAPEUTIC STRATEGY FOR IPN TREATMENT
- CONCLUSION AND PERSPECTIVES
- ACKNOWLEDGEMENTS
- CONFLICT OF INTEREST
- Figure
- Table
- Reference
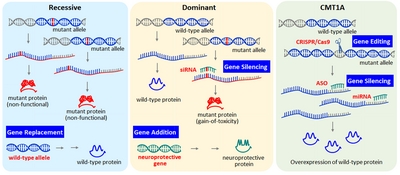
{kind=link}
Tables
- Go to
- Abstract
- Graphical Abstract
- INTRODUCTION
- MAJOR TARGETS FOR GENE THERAPY IN IPN
- GENE REPLACEMENT THERAPY
- GENE ADDITION THERAPY
- SUPPRESSION OF MUTANT GENE EXPRESSION
- TARGETING THE GENETIC DOSAGE BY POSTTRANSCRIPTIONAL MODULATION
- MODULATION OF TRANSCRIPTIONAL ACTIVITY
- OPTIMIZATION OF THERAPEUTIC STRATEGY FOR IPN TREATMENT
- CONCLUSION AND PERSPECTIVES
- ACKNOWLEDGEMENTS
- CONFLICT OF INTEREST
- Figure
- Table
- Reference
IPN causative genes and the inheritance pattern
| Type | Dominant | Recessive |
|---|---|---|
| Demyelinating type | PMP22, PMP2, MPZ*, LITAF, FBLN5, EGR2*, NEFL* | MPZ*, NEFL*, EGR2*, GDAP1*, MTMR2, SH3TC2, NDRG1, PRX, FGD4, SBF1, SBF2, FIG4, CTDP1, SURF1, ADCY6, CNTNAP1, HK1 |
| Axonal type | MFN2, GDAP1*, LRSAM1*, MPZ*, NEFH, KIF5A, ATP1A1, VCP, TFG, DHTKD1, TUBB3, NAGLU, DCAF8, DGAT2, MORC2, HSPB1, HSPB3, HSPB8, GARS, AARS, HARS, MARS, DYNC1H1, BICD2, REEP1, BSCL2, SETX, SLC5A7, MYH14, TRPV4, RAB7 | LMNA, PNKP, TRIM2, SPG11, MME, MCM3AP, SLC25A46, SCO2, MPV17, LRSAM1*, C12orf65, IGHMBP2, SIGMAR1, VRK1, ATP7A, UBA1, GLE1, LAS1L |
| Intermediate type (mixed type) | NEFL*, MPZ*, GJB1, YARS, INF2, DRP2, DNM2, GNB4, PDK3 | GDAP1*, COX6A1, PLEKHG5, KARS, AIFM1, PRPS1 |
| Incidence | > 95% | < 5% |
*Mutations in the gene cause dominant or recessive inheritance.
Gene therapies validated in animal models of IPN
| Type | Target | Therapeutic gene | Mode of action | Vector | Route | Reference |
|---|---|---|---|---|---|---|
| Gene addition | CMT1A | Neurotrophin-3 | Stimulation of neurite outgrowth and myelination | AAV | Intramuscular | 50 |
| Gene replacement | CMT4C | SH3TC2 | Replacement of the autosomal recessive gene | Lentivirus | Intrathecal | 36 |
| CMTX1 | GJB1 | Delivery of a wild-type gene | Lentivirus | Intraneural (sciatic nerve) | 41 | |
| Delivery of a wild-type gene | Lentivirus | Intrathecal | 42, 43 | |||
| Gene silencing | CMT1E | siRNA | Targeting and suppressing the mutant allele of PMP22 | Naked (synthetic siRNA) | Intraperitoneal | 61 |
| CMT1A | miRNA (miR-381) | Downregulating PMP22 overexpression | Lentivirus | Intraneural (sciatic nerve) | 69 | |
| Antisense oligonucleotide | Downregulating PMP22 overexpression by exon skipping | Naked (synthetic oligonucleotide) | Subcutaneous | 80 | ||
| Gene editing | CMT1A | CRISPR/Cas9 | Downregulating PMP22 overexpression by disrupting TATA-box | Naked (protein/gRNA) | Intraneural (sciatic nerve) | 82 |
Strategies to improve therapeutic efficacy for IPN treatment
| Type | Category | Strategy |
|---|---|---|
| Viral delivery | Toxicity | Reduction of genotoxicity using specific target sequence to avoid activation of proto-oncogene or using regulatory machinery for transgene expression |
| Efficiency | Enhancement of viral tropism specific to peripheral nervous system | |
| Non-viral delivery | Stability | Development of enhanced vehicle to increase the stability of oligonucleotides |
| Chemical modification of oligonucleotide to increase the stability | ||
| Efficiency | Development of novel chemical composition or peptide to enhance cell uptake and to facilitate endolysosomal escape or nuclear translocation | |
| Specificity | Isolation of novel receptors or membrane compositions in Schwann cell or axon | |
| Development of novel ligands specific to peripheral nervous system |
References
- Go to
- Abstract
- Graphical Abstract
- INTRODUCTION
- MAJOR TARGETS FOR GENE THERAPY IN IPN
- GENE REPLACEMENT THERAPY
- GENE ADDITION THERAPY
- SUPPRESSION OF MUTANT GENE EXPRESSION
- TARGETING THE GENETIC DOSAGE BY POSTTRANSCRIPTIONAL MODULATION
- MODULATION OF TRANSCRIPTIONAL ACTIVITY
- OPTIMIZATION OF THERAPEUTIC STRATEGY FOR IPN TREATMENT
- CONCLUSION AND PERSPECTIVES
- ACKNOWLEDGEMENTS
- CONFLICT OF INTEREST
- Figure
- Table
- Reference
- Harding AE, Thomas PK (1980) Genetic aspects of hereditary motor and sensory neuropathy (types I and II). J Med Genet 17: 329-336



- Klein CJ, Duan X, Shy ME (2013) Inherited neuropathies: clinical overview and update. Muscle Nerve 48: 604-622
- Sereda MW, Meyer zu Hörste G, Suter U, Uzma N, Nave KA (2003) Therapeutic administration of progesterone antagonist in a model of Charcot-Marie-Tooth disease (CMT-1A). Nat Med 9: 1533-1537
- Pareyson D, Reilly MM, Schenone A, Fabrizi GM, Cavallaro T, Santoro L, Vita G, Quattrone A, Padua L, Gemignani F, Visioli F, Laurà M, Radice D, Calabrese D, Hughes RA, Solari A; CMT-TRIAAL, CMT-TRAUKgroups (2011) Ascorbic acid in Charcot-Marie-Tooth disease type 1A (CMT-TRIAAL and CMT-TRAUK): a double-blind randomised trial. Lancet Neurol 10: 320-328
- Lewis RA, McDermott MP, Herrmann DN, Hoke A, Clawson LL, Siskind C, Feely SM, Miller LJ, Barohn RJ, Smith P, Luebbe E, Wu X, Shy ME; Muscle Study Group (2013) High-dosage ascorbic acid treatment in Charcot-Marie-Tooth disease type 1A: results of a randomized, double-masked, controlled trial. JAMA Neurol 70: 981-987
- Chumakov I, Milet A, Cholet N, Primas G, Boucard A, Pereira Y, Graudens E, Mandel J, Laffaire J, Foucquier J, Glibert F, Bertrand V, Nave KA, Sereda MW, Vial E, Guedj M, Hajj R, Nabirotchkin S, Cohen D (2014) Polytherapy with a combination of three repurposed drugs (PXT3003) down-regulates Pmp22 over-expression and improves myelination, axonal and functional parameters in models of CMT1A neuropathy. Orphanet J Rare Dis 9: 201
- Attarian S, Vallat JM, Magy L, Funalot B, Gonnaud PM, Lacour A, Péréon Y, Dubourg O, Pouget J, Micallef J, Franques J, Lefebvre MN, Ghorab K, Al-Moussawi M, Tiffreau V, Preudhomme M, Magot A, Leclair-Visonneau L, Stojkovic T, Bossi L, Lehert P, Gilbert W, Bertrand V, Mandel J, Milet A, Hajj R, Boudiaf L, Scart-Grès C, Nabirotchkin S, Guedj M, Chumakov I, Cohen D (2014) An exploratory randomised double-blind and placebo-controlled phase 2 study of a combination of baclofen, naltrexone and sorbitol (PXT3003) in patients with Charcot-Marie-Tooth disease type 1A. Orphanet J Rare Dis 9: 199
- Prukop T, Stenzel J, Wernick S, Kungl T, Mroczek M, Adam J, Ewers D, Nabirotchkin S, Nave KA, Hajj R, Cohen D, Sereda MW (2019) Early short-term PXT3003 combinational therapy delays disease onset in a transgenic rat model of Charcot-Marie-Tooth disease 1A (CMT1A). PLoS One 14: e0209752
- Lupski JR, de Oca-Luna RM, Slaugenhaupt S, Pentao L, Guzzetta V, Trask BJ, Saucedo-Cardenas O, Barker DF, Killian JM, Garcia CA, Chakravarti A, Patel PI (1991) DNA duplication associated with Charcot-Marie-Tooth disease type 1A. Cell 66: 219-232
- Patel PI, Roa BB, Welcher AA, Schoener-Scott R, Trask BJ, Pentao L, Snipes GJ, Garcia CA, Francke U, Shooter EM, Lupski JR, Suter U (1992) The gene for the peripheral myelin protein PMP-22 is a candidate for Charcot-Marie-Tooth disease type 1A. Nat Genet 1: 159-165
- Suter U, Welcher AA, Ozcelik T, Snipes GJ, Kosaras B, Francke U, Billings-Gagliardi S, Sidman RL, Shooter EM (1992) Trembler mouse carries a point mutation in a myelin gene. Nature 356: 241-244
- Valentijn LJ, Bolhuis PA, Zorn I, Hoogendijk JE, van den Bosch N, Hensels GW, Stanton VP Jr, Housman DE, Fischbeck KH, Ross DA, Nicholson GA, Meershoek EJ, Dauwerse HG, van Ommen GJB, Baas F (1992) The peripheral myelin gene PMP-22/GAS-3 is duplicated in Charcot-Marie-Tooth disease type 1A. Nat Genet 1: 166-170
- Eggermann K, Gess B, Häusler M, Weis J, Hahn A, Kurth I (2018) Hereditary neuropathies. Dtsch Arztebl Int 115: 91-97
- Pipis M, Rossor AM, Laura M, Reilly MM (2019) Next-generation sequencing in Charcot-Marie-Tooth disease: opportunities and challenges. Nat Rev Neurol 15: 644-656
- Bergamin G, Boaretto F, Briani C, Pegoraro E, Cacciavillani M, Martinuzzi A, Muglia M, Vettori A, Vazza G, Mostacciuolo ML (2014) Mutation analysis of MFN2, GJB1, MPZ and PMP22 in Italian patients with axonal Charcot-Marie-Tooth disease. Neuromolecular Med 16: 540-550
- Pareyson D, Saveri P, Pisciotta C (2017) New developments in Charcot-Marie-Tooth neuropathy and related diseases. Curr Opin Neurol 30: 471-480
- Saporta AS, Sottile SL, Miller LJ, Feely SM, Siskind CE, Shy ME (2011) Charcot-Marie-Tooth disease subtypes and genetic testing strategies. Ann Neurol 69: 22-33
- Murphy SM, Laura M, Fawcett K, Pandraud A, Liu YT, Davidson GL, Rossor AM, Polke JM, Castleman V, Manji H, Lunn MP, Bull K, Ramdharry G, Davis M, Blake JC, Houlden H, Reilly MM (2012) Charcot-Marie-Tooth disease: frequency of genetic subtypes and guidelines for genetic testing. J Neurol Neurosurg Psychiatry 83: 706-710
- Wang D, Gao G (2014) State-of-the-art human gene therapy: part II. Gene therapy strategies and clinical applications. Discov Med 18: 151-161
- McManus MT, Sharp PA (2002) Gene silencing in mammals by small interfering RNAs. Nat Rev Genet 3: 737-747
- Mali P, Esvelt KM, Church GM (2013) Cas9 as a versatile tool for engineering biology. Nat Methods 10: 957-963
- Piguet F, Alves S, Cartier N (2017) Clinical gene therapy for neurodegenerative diseases: past, present, and future. Hum Gene Ther 28: 988-1003
- Passage E, Norreel JC, Noack-Fraissignes P, Sanguedolce V, Pizant J, Thirion X, Robaglia-Schlupp A, Pellissier JF, Fontés M (2004) Ascorbic acid treatment corrects the phenotype of a mouse model of Charcot-Marie-Tooth disease. Nat Med 10: 396-401
- Khajavi M, Inoue K, Wiszniewski W, Ohyama T, Snipes GJ, Lupski JR (2005) Curcumin treatment abrogates endoplasmic reticulum retention and aggregation-induced apoptosis associated with neuropathy-causing myelin protein zero-truncating mutants. Am J Hum Genet 77: 841-850
- Rangaraju S, Madorsky I, Pileggi JG, Kamal A, Notterpek L (2008) Pharmacological induction of the heat shock response improves myelination in a neuropathic model. Neurobiol Dis 32: 105-115
- Takahashi R, Ikeda T, Hamaguchi A, Iwasa K, Yamada M (2012) Coenzyme Q10 therapy in hereditary motor sensory neuropathy type VI with novel mitofusin 2 mutation. Intern Med 51: 791-793
- Rocha AG, Franco A, Krezel AM, Rumsey JM, Alberti JM, Knight WC, Biris N, Zacharioudakis E, Janetka JW, Baloh RH, Kitsis RN, Mochly-Rosen D, Townsend RR, Gavathiotis E, Dorn GW 2nd (2018) MFN2 agonists reverse mitochondrial defects in preclinical models of Charcot-Marie-Tooth disease type 2A. Science 360: 336-341
- d'Ydewalle C, Krishnan J, Chiheb DM, Van Damme P, Irobi J, Kozikowski AP, Vanden Berghe P, Timmerman V, Robberecht W, Van Den Bosch L (2011) HDAC6 inhibitors reverse axonal loss in a mouse model of mutant HSPB1-induced Charcot-Marie-Tooth disease. Nat Med 17: 968-974
- Kim JY, Woo SY, Hong YB, Choi H, Kim J, Choi H, Mook-Jung I, Ha N, Kyung J, Koo SK, Jung SC, Choi BO (2016) HDAC6 inhibitors rescued the defective axonal mitochondrial movement in motor neurons derived from the induced pluripotent stem cells of peripheral neuropathy patients with HSPB1 mutation. Stem Cells Int 2016: 9475981
- Benoy V, Vanden Berghe P, Jarpe M, Van Damme P, Robberecht W, Van Den Bosch L (2017) Development of improved HDAC6 inhibitors as pharmacological therapy for axonal Charcot-Marie-Tooth disease. Neurotherapeutics 14: 417-428
- Benoy V, Van Helleputte L, Prior R, d'Ydewalle C, Haeck W, Geens N, Scheveneels W, Schevenels B, Cader MZ, Talbot K, Kozikowski AP, Vanden Berghe P, Van Damme P, Robberecht W, Van Den Bosch L (2018) HDAC6 is a therapeutic target in mutant GARS-induced Charcot-Marie-Tooth disease. Brain 141: 673-687
- Burns J, Ouvrier RA, Yiu EM, Joseph PD, Kornberg AJ, Fahey MC, Ryan MM (2009) Ascorbic acid for Charcot-Marie-Tooth disease type 1A in children: a randomised, double-blind, placebo-controlled, safety and efficacy trial. Lancet Neurol 8: 537-544
- Meyer zu Horste G, Prukop T, Liebetanz D, Mobius W, Nave KA, Sereda MW (2007) Antiprogesterone therapy uncouples axonal loss from demyelination in a transgenic rat model of CMT1A neuropathy. Ann Neurol 61: 61-72
- Zhou Y, Carmona S, Muhammad AKMG, Bell S, Landeros J, Vazquez M, Ho R, Franco A, Lu B, Dorn GW 2nd, Wang S, Lutz CM, Baloh RH (2019) Restoring mitofusin balance prevents axonal degeneration in a Charcot-Marie-Tooth type 2A model. J Clin Invest 130: 1756-1771
- Colby J, Nicholson R, Dickson KM, Orfali W, Naef R, Suter U, Snipes GJ (2000) PMP22 carrying the trembler or trembler-J mutation is intracellularly retained in myelinating Schwann cells. Neurobiol Dis 7(6 Pt B): 561-573
- Schiza N, Georgiou E, Kagiava A, Médard JJ, Richter J, Tryfonos C, Sargiannidou I, Heslegrave AJ, Rossor AM, Zetterberg H, Reilly MM, Christodoulou C, Chrast R, Kleopa KA (2019) Gene replacement therapy in a model of Charcot-Marie-Tooth 4C neuropathy. Brain 142: 1227-1241
- Senderek J, Bergmann C, Stendel C, Kirfel J, Verpoorten N, De Jonghe P, Timmerman V, Chrast R, Verheijen MH, Lemke G, Battaloglu E, Parman Y, Erdem S, Tan E, Topaloglu H, Hahn A, Müller-Felber W, Rizzuto N, Fabrizi GM, Stuhrmann M, Rudnik-Schöneborn S, Züchner S, Michael Schröder J, Buchheim E, Straub V, Klepper J, Huehne K, Rautenstrauss B, Büttner R, Nelis E, Zerres K (2003) Mutations in a gene encoding a novel SH3/TPR domain protein cause autosomal recessive Charcot-Marie-Tooth type 4C neuropathy. Am J Hum Genet 73: 1106-1119
- Bergoffen J, Scherer SS, Wang S, Scott MO, Bone LJ, Paul DL, Chen K, Lensch MW, Chance PF, Fischbeck KH (1993) Connexin mutations in X-linked Charcot-Marie-Tooth disease. Science 262: 2039-2042
- Scherer SS, Deschênes SM, Xu YT, Grinspan JB, Fischbeck KH, Paul DL (1995) Connexin32 is a myelin-related protein in the PNS and CNS. J Neurosci 15: 8281-8294
- Hong YB, Park JM, Yu JS, Yoo DH, Nam DE, Park HJ, Lee JS, Hwang SH, Chung KW, Choi BO (2017) Clinical characterization and genetic analysis of Korean patients with X-linked Charcot-Marie-Tooth disease type 1. J Peripher Nerv Syst 22: 172-181
- Sargiannidou I, Kagiava A, Bashiardes S, Richter J, Christodoulou C, Scherer SS, Kleopa KA (2015) Intraneural GJB1 gene delivery improves nerve pathology in a model of X-linked Charcot-Marie-Tooth disease. Ann Neurol 78: 303-316
- Kagiava A, Sargiannidou I, Theophilidis G, Karaiskos C, Richter J, Bashiardes S, Schiza N, Nearchou M, Christodoulou C, Scherer SS, Kleopa KA (2016) Intrathecal gene therapy rescues a model of demyelinating peripheral neuropathy. Proc Natl Acad Sci U S A 113: E2421-E2429
- Kagiava A, Richter J, Tryfonos C, Karaiskos C, Heslegrave AJ, Sargiannidou I, Rossor AM, Zetterberg H, Reilly MM, Christodoulou C, Kleopa KA (2019) Gene replacement therapy after neuropathy onset provides therapeutic benefit in a model of CMT1X. Hum Mol Genet 28: 3528-3542
- Huang EJ, Reichardt LF (2001) Neurotrophins: roles in neuronal development and function. Annu Rev Neurosci 24: 677-736
- Woolley AG, Tait KJ, Hurren BJ, Fisher L, Sheard PW, Duxson MJ (2008) Developmental loss of NT-3 in vivo results in reduced levels of myelin-specific proteins, a reduced extent of myelination and increased apoptosis of Schwann cells. Glia 56: 306-317
- Zhang JY, Luo XG, Xian CJ, Liu ZH, Zhou XF (2000) Endogenous BDNF is required for myelination and regeneration of injured sciatic nerve in rodents. Eur J Neurosci 12: 4171-4180
- Meier C, Parmantier E, Brennan A, Mirsky R, Jessen KR (1999) Developing Schwann cells acquire the ability to survive without axons by establishing an autocrine circuit involving insulin-like growth factor, neurotrophin-3, and platelet-derived growth factor-BB. J Neurosci 19: 3847-3859
- Sahenk Z, Nagaraja HN, McCracken BS, King WM, Freimer ML, Cedarbaum JM, Mendell JR (2005) NT-3 promotes nerve regeneration and sensory improvement in CMT1A mouse models and in patients. Neurology 65: 681-689
- Sahenk Z, Galloway G, Edwards C, Malik V, Kaspar BK, Eagle A, Yetter B, Forgie A, Tsao D, Lin JC (2010) TrkB and TrkC agonist antibodies improve function, electrophysiologic and pathologic features in Trembler J mice. Exp Neurol 224: 495-506
- Sahenk Z, Galloway G, Clark KR, Malik V, Rodino-Klapac LR, Kaspar BK, Chen L, Braganza C, Montgomery C, Mendell JR (2014) AAV1.NT-3 gene therapy for Charcot-Marie-Tooth neuropathy. Mol Ther 22:511-521.
- Fledrich R, Stassart RM, Klink A, Rasch LM, Prukop T, Haag L, Czesnik D, Kungl T, Abdelaal TA, Keric N, Stadelmann C, Brück W, Nave KA, Sereda MW (2014) Soluble neuregulin-1 modulates disease pathogenesis in rodent models of Charcot-Marie-Tooth disease 1A. Nat Med 20: 1055-1061
- Belin S, Ornaghi F, Shackleford G, Wang J, Scapin C, Lopez-Anido C, Silvestri N, Robertson N, Williamson C, Ishii A, Taveggia C, Svaren J, Bansal R, Schwab MH, Nave K, Fratta P, D'Antonio M, Poitelon Y, Feltri ML, Wrabetz L (2019) Neuregulin 1 type III improves peripheral nerve myelination in a mouse model of congenital hypomyelinating neuropathy. Hum Mol Genet 28: 1260-1273
- Chi X, Gatti P, Papoian T (2017) Safety of antisense oligonucleotide and siRNA-based therapeutics. Drug Discov Today 22: 823-833
- Fellmann C, Lowe SW (2014) Stable RNA interference rules for silencing. Nat Cell Biol 16: 10-18
- Moore CB, Guthrie EH, Huang MT, Taxman DJ (2010) Short hairpin RNA (shRNA): design, delivery, and assessment of gene knockdown. Methods Mol Biol 629: 141-158
- Feng X, Zhao P, He Y, Zuo Z (2006) Allele-specific silencing of Alzheimer's disease genes: the amyloid precursor protein genes with Swedish or London mutations. Gene 371: 68-74
- Takahashi M, Suzuki M, Fukuoka M, Fujikake N, Watanabe S, Murata M, Wada K, Nagai Y, Hohjoh H (2015) Normalization of overexpressed α-synuclein causing Parkinson's disease by a moderate gene silencing with RNA interference. Mol Ther Nucleic Acids 4: e241
- Carroll JB, Warby SC, Southwell AL, Doty CN, Greenlee S, Skotte N, Hung G, Bennett CF, Freier SM, Hayden MR (2011) Potent and selective antisense oligonucleotides targeting single-nucleotide polymorphisms in the Huntington disease gene / allele-specific silencing of mutant huntingtin. Mol Ther 19: 2178-2185
- Nóbrega C, Nascimento-Ferreira I, Onofre I, Albuquerque D, Hirai H, Déglon N, de Almeida LP (2013) Silencing mutant ataxin-3 rescues motor deficits and neuropathology in Machado-Joseph disease transgenic mice. PLoS One 8: e52396
- Xia X, Zhou H, Huang Y, Xu Z (2006) Allele-specific RNAi selectively silences mutant SOD1 and achieves significant therapeutic benefit in vivo. Neurobiol Dis 23: 578-586
- Lee JS, Chang EH, Koo OJ, Jwa DH, Mo WM, Kwak G, Moon HW, Park HT, Hong YB, Choi BO (2017) Pmp22 mutant allele-specific siRNA alleviates demyelinating neuropathic phenotype in vivo. Neurobiol Dis 100: 99-107
- Jang SW, Lopez-Anido C, MacArthur R, Svaren J, Inglese J (2012) Identification of drug modulators targeting gene-dosage disease CMT1A. ACS Chem Biol 7: 1205-1213
- Bartel DP (2004) MicroRNAs: genomics, biogenesis, mechanism, and function. Cell 116: 281-297
- Bremer J, O'Connor T, Tiberi C, Rehrauer H, Weis J, Aguzzi A (2010) Ablation of Dicer from murine Schwann cells increases their proliferation while blocking myelination. PLoS One 5(8): e12450
- Pereira JA, Baumann R, Norrmen C, Somandin C, Miehe M, Jacob C, Luhmann T, Hall-Bozic H, Mantei N, Meijer D, Suter U (2010) Dicer in Schwann cells is required for myelination and axonal integrity. J Neurosci 30: 6763-6775
- Yun B, Anderegg A, Menichella D, Wrabetz L, Feltri ML, Awatramani R (2010) MicroRNA-deficient Schwann cells display congenital hypomyelination. J Neurosci 30: 7722-7728
- Verrier JD, Semple-Rowland S, Madorsky I, Papin JE, Notterpek L (2010) Reduction of Dicer impairs Schwann cell differentiation and myelination. J Neurosci Res 88: 2558-2568
- Verrier JD, Lau P, Hudson L, Murashov AK, Renne R, Notterpek L (2009) Peripheral myelin protein 22 is regulated post-transcriptionally by miRNA-29a. Glia 57: 1265-1279
- Lee JS, Kwak G, Kim HJ, Park HT, Choi BO, Hong YB (2019) miR-381 attenuates peripheral neuropathic phenotype caused by overexpression of PMP22. Exp Neurobiol 28: 279-288
- Huxley C, Passage E, Manson A, Putzu G, Figarella-Branger D, Pellissier JF, Fontés M (1996) Construction of a mouse model of Charcot-Marie-Tooth disease type 1A by pronuclear injection of human YAC DNA. Hum Mol Genet 5: 563-569
- Huxley C, Passage E, Robertson AM, Youl B, Huston S, Manson A, Sabéran-Djoniedi D, Figarella-Branger D, Pellissier JF, Thomas PK, Fontés M (1998) Correlation between varying levels of PMP22 expression and the degree of demyelination and reduction in nerve conduction velocity in transgenic mice. Hum Mol Genet 7: 449-458
- Chittoor VG, Sooyeon L, Rangaraju S, Nicks JR, Schmidt JT, Madorsky I, Narvaez DC, Notterpek L (2013) Biochemical characterization of protein quality control mechanisms during disease progression in the C22 mouse model of CMT1A. ASN Neuro 5: e00128
- Bennett CF, Swayze EE (2010) RNA targeting therapeutics: molecular mechanisms of antisense oligonucleotides as a therapeutic platform. Annu Rev Pharmacol Toxicol 50: 259-293
- Rigo F, Hua Y, Krainer AR, Bennett CF (2012) Antisense-based therapy for the treatment of spinal muscular atrophy. J Cell Biol 199: 21-25
- Finkel RS, Chiriboga CA, Vajsar J, Day JW, Montes J, De Vivo DC, Yamashita M, Rigo F, Hung G, Schneider E, Norris DA, Xia S, Bennett CF, Bishop KM (2016) Treatment of infantile-onset spinal muscular atrophy with nusinersen: a phase 2, open-label, dose-escalation study. Lancet 388: 3017-3026
- Mercuri E, Darras BT, Chiriboga CA, Day JW, Campbell C, Connolly AM, Iannaccone ST, Kirschner J, Kuntz NL, Saito K, Shieh PB, Tulinius M, Mazzone ES, Montes J, Bishop KM, Yang Q, Foster R, Gheuens S, Bennett CF, Farwell W, Schneider E, De Vivo DC, Finkel RS; CHERISH Study Group (2018) Nusinersen versus Sham control in later-onset spinal muscular atrophy. N Engl J Med 378: 625-635
- Mann CJ, Honeyman K, Cheng AJ, Ly T, Lloyd F, Fletcher S, Morgan JE, Partridge TA, Wilton SD (2001) Antisense-induced exon skipping and synthesis of dystrophin in the mdx mouse. Proc Natl Acad Sci U S A 98: 42-47
- Cirak S, Arechavala-Gomeza V, Guglieri M, Feng L, Torelli S, Anthony K, Abbs S, Garralda ME, Bourke J, Wells DJ, Dickson G, Wood MJ, Wilton SD, Straub V, Kole R, Shrewsbury SB, Sewry C, Morgan JE, Bushby K, Muntoni F (2011) Exon skipping and dystrophin restoration in patients with Duchenne muscular dystrophy after systemic phosphorodiamidate morpholino oligomer treatment: an open-label, phase 2, dose-escalation study. Lancet 378: 595-605
- Monani UR, Lorson CL, Parsons DW, Prior TW, Androphy EJ, Burghes AH, McPherson JD (1999) A single nucleotide difference that alters splicing patterns distinguishes the SMA gene SMN1 from the copy gene SMN2. Hum Mol Genet 8: 1177-1183
- Zhao HT, Damle S, Ikeda-Lee K, Kuntz S, Li J, Mohan A, Kim A, Hung G, Scheideler MA, Scherer SS, Svaren J, Swayze EE, Kordasiewicz HB (2018) PMP22 antisense oligonucleotides reverse Charcot-Marie-Tooth disease type 1A features in rodent models. J Clin Invest 128: 359-368
- Pantera H, Moran JJ, Hung HA, Pak E, Dutra A, Svaren J (2018) Regulation of the neuropathy-associated Pmp22 gene by a distal super-enhancer. Hum Mol Genet 27: 2830-2839
- Lee JS, Lee JY, Song DW, Bae HS, Doo HM, Yu HS, Lee KJ, Kim HK, Hwang H, Kwak G, Kim D, Kim S, Hong YB, Lee JM, Choi BO (2020) Targeted PMP22 TATA-box editing by CRISPR/Cas9 reduces demyelinating neuropathy of Charcot-Marie-Tooth disease type 1A in mice. Nucleic Acids Res 48: 130-140
- Zhang F, Wen Y, Guo X (2014) CRISPR/Cas9 for genome editing: progress, implications and challenges. Hum Mol Genet 23: R40-R46
- Molla KA, Yang Y (2019) CRISPR/Cas-mediated base editing: technical considerations and practical applications. Trends Biotechnol 37: 1121-1142
- David RM, Doherty AT (2017) Viral vectors: the road to reducing genotoxicity. Toxicol Sci 155: 315-325
- Niederer HA, Bangham CR (2014) Integration site and clonal expansion in human chronic retroviral infection and gene therapy. Viruses 6: 4140-4164
- Emery DW (2011) The use of chromatin insulators to improve the expression and safety of integrating gene transfer vectors. Hum Gene Ther 22: 761-774
- Zhong G, Wang H, He W, Li Y, Mou H, Tickner ZJ, Tran MH, Ou T, Yin Y, Diao H, Farzan M (2020) A reversible RNA on-switch that controls gene expression of AAV-delivered therapeutics in vivo. Nat Biotechnol 38: 169-175
- Hoyng SA, De Winter F, Gnavi S, van Egmond L, Attwell CL, Tannemaat MR, Verhaagen J, Malessy MJ (2015) Gene delivery to rat and human Schwann cells and nerve segments: a comparison of AAV 1-9 and lentiviral vectors. Gene Ther 22: 767-780
- Homs J, Ariza L, Pagès G, Udina E, Navarro X, Chillón M, Bosch A (2011) Schwann cell targeting via intrasciatic injection of AAV8 as gene therapy strategy for peripheral nerve regeneration. Gene Ther 18: 622-630
- Wu P, Chen H, Jin R, Weng T, Ho JK, You C, Zhang L, Wang X, Han C (2018) Non-viral gene delivery systems for tissue repair and regeneration. J Transl Med 16: 29
- Hu Q, Katti PS, Gu Z (2014) Enzyme-responsive nanomaterials for controlled drug delivery. Nanoscale 6: 12273-12286
- Lai WF, Wong WT (2018) Design of polymeric gene carriers for effective intracellular delivery. Trends Biotechnol 36: 713-728
- Nuzzo S, Roscigno G, Affinito A, Ingenito F, Quintavalle C, Condorelli G (2019) Potential and challenges of aptamers as specific carriers of therapeutic oligonucleotides for precision medicine in cancer. Cancers (Basel) 11: E1521