Articles

Article Tools
 View Full Text View Full Text |
 Abstract Abstract |
 Article as PDF Article as PDF |
 Print this Article Print this Article |
 Pubmed Pubmed |
 PMC PMC |
 PubReader PubReader |
 Export to Citation Export to Citation |
 Email Alerts Email Alerts |
 Open Access Open Access |
Share this article on :



Stats or Metrics
Article
Original Article
Exp Neurobiol 2021; 30(1): 59-72
Published online January 19, 2021
https://doi.org/10.5607/en20055
© The Korean Society for Brain and Neural Sciences
Regulation of Gut Microbiota Disrupts the Glucocorticoid Receptor Pathway and Inflammation-related Pathways in the Mouse Hippocampus
Xuechen Rao1,2†, Lanxiang Liu2,3†, Haiyang Wang2,4†, Ying Yu2†, Wenxia Li2, Tingjia Chai2, Wei Zhou2, Ping Ji4, Jinlin Song4, Hong Wei5* and Peng Xie2,6*
1College of Biomedical Engineering, Chongqing Medical University, Chongqing 400016, 2NHC Key Laboratory of Diagnosis and Treatment on Brain Functional Diseases, The First Affiliated Hospital of Chongqing Medical University, Chongqing 400016, 3Department of Neurology, Yongchuan Hospital of Chongqing Medical University, Chongqing 402160, 4College of Stomatology and Affiliated Stomatological Hospital of Chongqing Medical University, Chongqing 401147, 5Department of Laboratory Animal Science, College of Basic Medical Sciences, Third Military Medical University, Chongqing 400038, 6Department of Neurology, The First Affiliated Hospital of Chongqing Medical University, Chongqing 400016, China
Correspondence to: *To whom correspondence should be addressed.
Peng Xie, TEL: 86-023-68485490, FAX: 86-023-68485111
e-mail: xiepeng@cqmu.edu.cn
Hong Wei, TEL: 86-023-68752051, FAX: 86-023-68752051
e-mail: weihong63528@163.com
†These authors contributed equally to this work.
This is an Open Access article distributed under the terms of the Creative Commons Attribution Non-Commercial License (http://creativecommons.org/licenses/by-nc/4.0) which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
Abstract
An increasing number of studies have recently indicated the important effects of gut microbes on various functions of the central nervous system. However, the underlying mechanisms by which gut microbiota regulate brain functions and behavioral phenotypes remain largely unknown. We therefore used isobaric tags for relative and absolute quantitation (iTRAQ)-based quantitative proteomic analysis to obtain proteomic profiles of the hippocampus in germ-free (GF), colonized GF, and specific pathogen-free (SPF) mice. We then integrated the resulting proteomic data with previously reported mRNA microarray data, to further explore the effects of gut microbes on host brain functions. We identified that 61 proteins were upregulated and 242 proteins were downregulated in GF mice compared with SPF mice. Of these, 124 proteins were significantly restored following gut microbiota colonization. Bioinformatic analysis of these significant proteins indicated that the glucocorticoid receptor signaling pathway and inflammation-related pathways were the most enriched disrupted pathways. This study provides new insights into the pathological mechanisms of gut microbiota-regulated diseases.
Graphical Abstract
Keywords: Germ-free, Gut microbiota, Proteomics, Hippocampus, Glucocorticoid receptor pathway, Inflammation-related pathways
INTRODUCTION
The human gastrointestinal tract contains approximately one trillion microorganisms, most of which are bacteria, belonging to more than 1,000 bacterial species [1]. Recent studies have suggested that the gut microbiota can have dramatic effects on the development and function of the host brain [2]. Moreover, accumulating studies have revealed bidirectional communication between the gut microbiota and the brain, and have proposed a novel conceptual model of a “microbiota–gut–brain axis” [3-5]. Through this axis, the gut and the brain may communicate via multiple mechanisms, including immune responses, the vagus nerve, short-chain fatty acids, endocrine signaling, and tryptophan metabolism [2, 6]. Many studies have found a high prevalence of mood disorders, such as depression and/or anxiety, in patients with gastrointestinal disorders including irritable bowel syndrome and inflammatory bowel disease [7, 8]. In our previous studies, we demonstrated the effects of the gut microbiota on brain function. For example, gut microbiota may modulate depression- and anxiety-like behaviors in mice through the hypothalamic–pituitary–adrenal (HPA) axis [9], and alter hippocampal RNA regulation and lipid metabolism [10, 11]. Furthermore, colonization of gut microbiota early in life might facilitate neurodevelopment via protein kinase C–cAMP response element-binding protein (CREB) signaling [12]. Sex-specific differences in gut microbiota composition in patients with major depressive disorder (MDD) were identified [13]. In general, disturbances in gut–brain axis may be highly correlated with pathophysiology of mood disorders.
Germ-free (GF) mice, who are born and raised without any exposure to microbes, allow us to better understand the effects of gut microbes on the host brain. Our previous studies have found that the GF mice exhibited antidepressant-like and anti-anxiety-like behaviors when comparing with SPF mice [10, 12, 14, 15]. Although colonized GF (CGF) mice–a complementary approach to explore the potential effects of the gut microbiota on brain function–showed no normalization of behavioral changes, the expression levels of numerous lncRNAs, miRNAs and mRNAs were significantly restored [11, 16, 17]. In addition, when transplanting fecal samples from patients with major depressive disorder and healthy controls to GF mice, respectively, the “depression microbiota” received mice showed significant depressive- and anxiety-like behaviors compared with “healthy microbiota” received mice [15], suggesting a causative role of gut microbiota dysbiosis in the onset of depression. However, the mechanism of gut microbiota drives anxiety- and depression-like behaviors is complex and still unclear. In the present study, we therefore focused our attention on the hippocampus, which is a crucial brain region that has for decades been recognized as critical in the pathogenesis of depression [18].
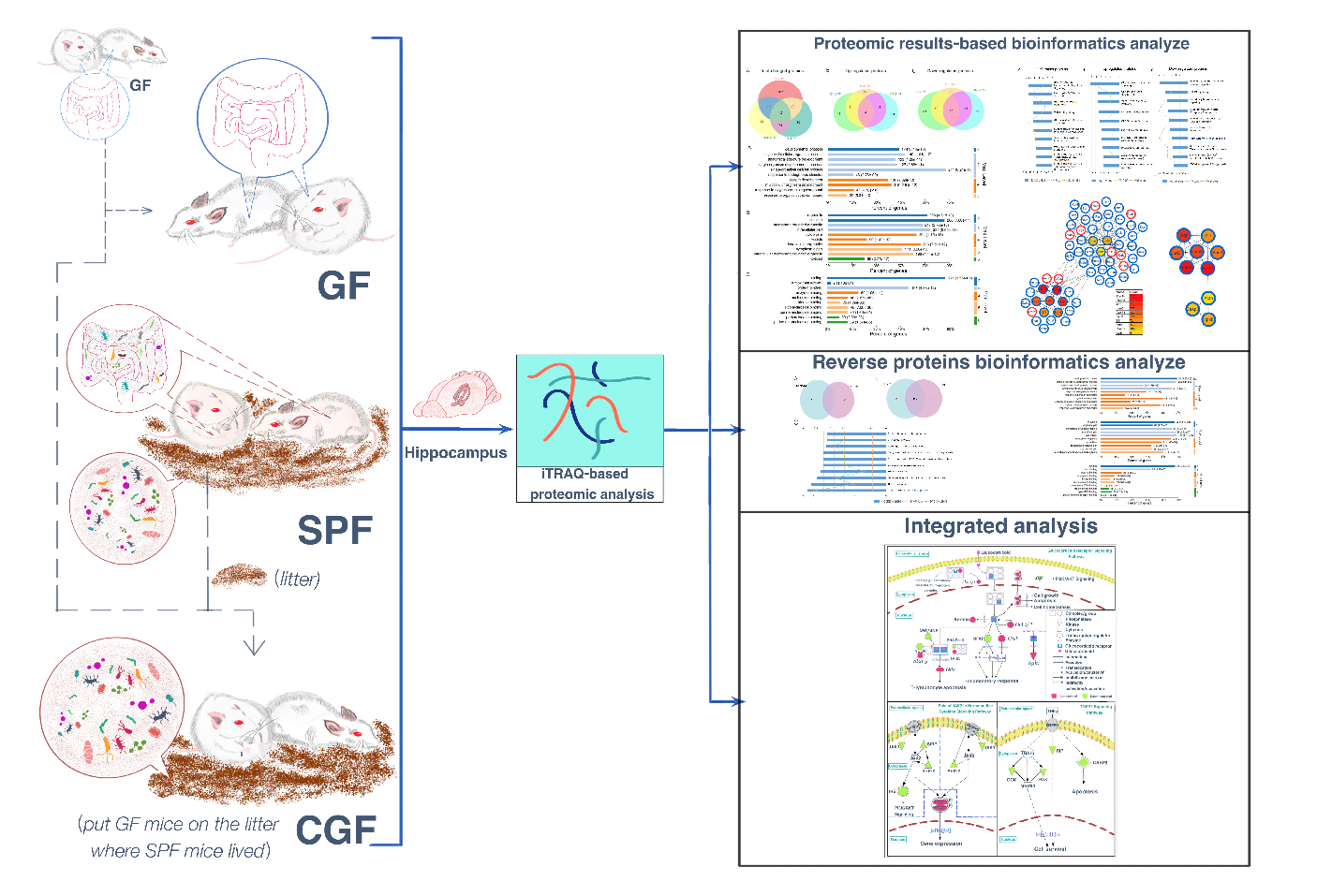
Isobaric tags for relative and absolute quantitation (iTRAQ) is a relatively practical protein detection method with high accuracy, sensitivity, and reproducibility. This method can be used to identify differentially expressed proteins in biological samples under different pathological conditions that are potential to uncover the underlying mechanisms of diseases. Using this approach, we have previously identified numerous differentially expressed proteins in the olfactory bulb of the gut microbiota-remodeled mice model of depression [19], and in the hippocampus of the chronic social defeat stress mice model, and identified the potential targets for depression treatment [20]. To investigate the underlying mechanisms by which gut microbes influence the depression-related brain function and behaviors, we used iTRAQ-based quantitative proteomic analysis to obtain proteomic profiles of the hippocampus of GF, CGF, and SPF mice, followed by the bioinformatics analysis. In addition, we integrated our current proteomic data with previously reported mRNA microarray data from GF vs. SPF mice [14] to further explore the underlying mechanisms.
MATERIALS AND METHODS
Animals
Nine-week-old male GF and SPF (
Sample collection and preparation
The mice were anesthetized with 10% chloral hydrate (400 mg/kg) followed by perfusion, and killed in random order by cervical dislocation [9]. The brain was quickly removed from the cranium, and the hippocampus was immediately stripped out and frozen in liquid nitrogen, and then stored at -80°C until used in the assay. To avoid any circadian impact on the results, all mouse brains were collected at the same time of day (between 09:00 and 11:00) [22]. For processing, samples were first subjected to liquid nitrogen grinding. Subsequently, each sample was dissolved and adequately homogenized in 1 ml of SDT buffer. All of the sample lysates were then sonicated on an ice-water mixture at 80 W for 7 s, followed by a pause for 8 s, which was repeated six times. After ultrasonic extraction, the lysates were centrifuged at 14,000×
iTRAQ labeling and SCX fraction
This part of the experiment was performed in a similar manner to that of previous studies [23, 24]. In line with the manufacturer’s instructions, pooled samples were extracted and digested, and the tryptic peptides were labeled using the iTRAQ Reagent-8PLEX Multiplex Kit. The samples were labeled with different iTRAQ tags. Subsequently, the labeled peptides from each group were mixed at equal ratios, and SCX chromatography was then used for further fractionation.
Liquid chromatography (LC)-mass spectrometry (MS)/MS and data analysis
The SCX fractions were subjected to LC-MS analysis and separated using a Thermo Easy-nLC binary buffer system. The composition of the buffer was buffer A (0.1% formic acid) and buffer B (consisting of 84% acetonitrile and 0.1% formic acid). The samples were first loaded into reversed-phase columns (20 mm×100 μm, 5 μm-C18). Next, analytical columns (75 μm×100 mm, 3 μm-C18) were used to separate the mixed peptides at a flow rate of 300 nl/minute for 60 minutes. The specific settings of the liquid-phase linear gradient was set as follows: minutes 0~55 with buffer B from 0% to 55%; minutes 55~57 with buffer B from 55% to 100%; and then buffer B maintained at 100% during minutes 57~60. Subsequently, each specimen was isolated by capillary high-performance LC and applied to a Q-Exactive mass spectrometer for protein identification. The specific detection method was as follows. The positive ion and parent ion were scanned in the range of 300 to 1,800 m/z. The first resolution of MS was 70,000 at m/z 200, and the secondary resolution of MS was 17,500 at m/z 200. The maximum ion injection times were fixed at 10 and 60 ms, respectively. After a full MS scan, the MS data required for further analysis were extracted from the top 10 most abundant precursor ions. The underfill ratio was 0.1% and the normalized collision energy was 30 eV. The protein identification and quantitative analysis methods were performed using Mascot 2.2 and Proteome Discoverer 1.4 software based on the UniProt (uniprot_Mouse_77271_20150609) database. The Mascot search parameters were set as reported in a previous study [24].
Bioinformatics analysis
For further analysis, the significantly differentially expressed proteins were selected using the following thresholds: p<0.05, fold change >1.2 or <0.83. To explore the functions of the differentially expressed proteins, we first used Gene Ontology (GO) enrichment analysis, which was performed using OmicsBean [25]. This describes the properties of genes and gene products in an organism over three aspects: biological process (BP), cellular component (CC), and molecular function (MF). We used Ingenuity Pathway Analysis (IPA, Ingenuity Systems, Redwood City, CA, USA; www.ingenuity.com) to identify significant dysfunctional canonical pathways and networks to obtain a better understanding of the pathophysiological effects of differentially expressed proteins on host [26]. We then mapped the differentially expressed proteins onto the Search Tool for the Retrieval of Interacting Genes/Proteins (STRING) database (v11.0; https://string-db.org/) to construct the protein–protein interactions (PPI) network. The PPI network was visualized by Cytoscape (v3.7.2), and the hub proteins were analyzed using the CytoHubba plug-in [27-29]. In addition, we integrated our present proteomic data with previously reported mRNA microarray data [14] to reveal the mechanisms by which the gut microbiota regulates brain functions and behavioral phenotypes.
RESULTS
Comparative proteome analysis
We used the well-established iTRAQ-based strategy to investigate proteomic changes in the hippocampus of GF, SPF, and CGF mice. The quantified protein sequence information was extracted in batches from the UniProtKB database. A total of 5,514 proteins were acquired. Based on the thresholds described above, numerous proteins were identified to be differentially expressed in GF vs. SPF, CGF vs. SPF, and CGF vs. GF comparisons, respectively. We performed an overlapping analysis of the differentially expressed proteins among these three comparison groups, and found that 42 common proteins were differentially expressed across the three comparisons (Fig. 1A). However, only 4 upregulated proteins (Fig. 1B) and 17 downregulated proteins (Fig. 1C) were identified with concordant direction of fold change across the three comparisons.
Functional analysis of differentially expressed proteins in the absence of gut microbiota
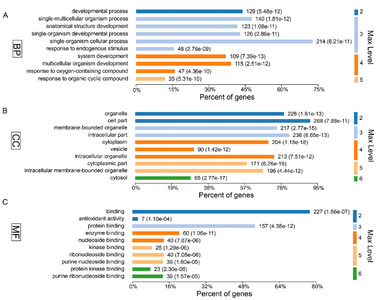
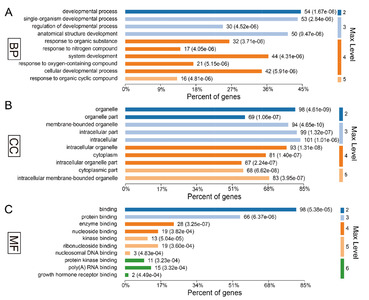
In present study, we found that the absence of gut microbiota results in significant changes in hippocampal protein expression in GF mice. Of the acquired 5,514 proteins, a total of 61 proteins were identified upregulation and 242 were downregulation in the GF mice compared with the SPF mice (Supplementary Table 1). To explore the biological functions of these significant proteins, we firstly performed GO annotations for these differentially expressed proteins. As a result, the response to organic cyclic compound was the most significantly annotated term in BP category at level 5 (p-value=5.31E-10, Fig. 2A), most of these proteins were annotated as cytosolic proteins in CC category (p-value=2.77E-17, Fig. 2B), and protein kinase binding was the most significant related function in MF category (p-value=2.30E-06, Fig. 2C).
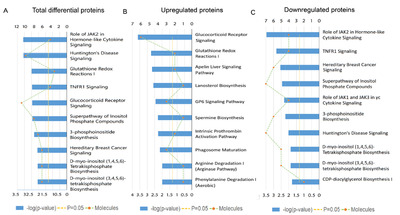
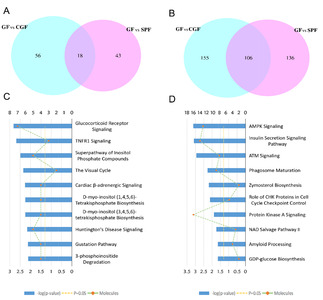
To get a better understanding of underlying mechanisms by which the absence of gut microbiota regulates the brain functions and behaviors of host. We performed functional enrichment pathway analysis for all these significant proteins, as well as the upregulated and downregulated proteins, respectively, using IPA software. The results revealed that the role of Janus kinase 2 (JAK2) in hormone-like cytokine signaling (p-value=8.13E-04), Huntington’s disease signaling (p-value=8.51E-04), glutathione redox reaction I (p-value=3.16E-03), Tumor necrosis factor receptor 1 (TNFR1) signaling (p-value=3.47E-03), and glucocorticoid receptor signaling (p-value=3.55E-03) were the top five disturbed functional pathways (Fig. 3A). However, the upregulated proteins were primarily enriched in the glucocorticoid receptor signaling pathway (p-value=2.29E-04, Fig. 3B), and the downregulated proteins were mainly involved in the role of JAK2 in hormone-like cytokine signaling and in TNFR1 signaling pathways (p-value=3.39E-04 and p-value=1.51E-03, respectively, Fig. 3C).
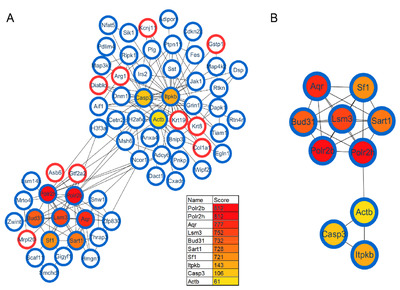
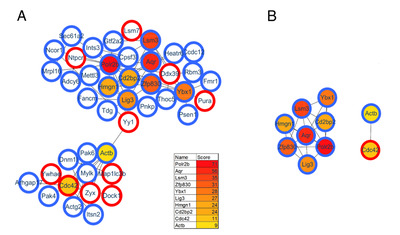
Protein interactions play an important role in regulating cells and their signaling pathways. We thus performed the PPI network analysis to further understand the interactions among these differentially expressed proteins in GF vs. SPF mice. As a result, a total of 282 significant proteins were annotated in STRING database. Using the CytoHubba plug-in, ten top-rank hub proteins were identified, and the PPI network of these hub proteins with their directly interacted proteins were shown in Fig. 4A, and that of the ten hub proteins per se in Fig. 4B. Of these hub proteins, POLR2B and POLR2H are the subunits of DNA-dependent RNA polymerase II, and play critical roles in the transcription. Interestingly, AQR, LSM3, BUD31, SART1 and SF1 are important components of spliceosome that catalyzes the pre-mRNA splicing. The downregulations of these spliceosome related proteins in GF mice comparted with the SPF mice, suggesting the inhibition of pre-mRNA alternative splicing, this findings was consistent with that of our previous study focused on the phosphorylation modification changes in the hippocampus of GF mice [30].
Functional analysis of proteins restored following gut microbiota colonization
To further explore the effects of gut microbiota on the protein expression of host. We analyzed the restored proteins following gut microbiota colonization that were characterized by their expression in opposite directions when examined in GF vs. SPF and CGF vs. GF comparisons, and were recognized as microbiota-specific proteins. As a result, a total of 124 proteins were significantly restored after gut microbiota colonization. Of all these 124 restored proteins, 18 were upregulated in GF mice compared with SPF mice, and 106 were downregulated. The expression levels of these restored proteins in the GF, SPF, and CGF mice are displayed in Supplementary Table 2. In the GO annotation analysis, these restored proteins were primarily related to the response to organic cyclic compounds in BP category at level 5 (p-value=4.81E-06, Fig. 5A), annotated in intracellular membrane-bound organelles in CC category at level 5 (p-value=3.05E-07, Fig. 5B), and had protein kinase binding functions in MF category at level 6 (p-value=3.23E-04, Fig. 5C). Moreover, we performed functional pathway analysis using IPA software to further explore the biological functions of these 124 microbiota-specific proteins (Fig. 6A and 6B), and found that glucocorticoid receptor signaling (p-value=1.51E-03) and TNFR1 signaling (p-value=2.04E-03) pathways were the primarily enriched disrupted pathways (Fig. 6C). This was consistent with the results found in mice with an absence of gut microbiota, as described in the previous section.
As a complementary approach to evaluate the restored protein-related pathways after gut microbiota colonization, we performed functional analysis of differentially expressed proteins in GF vs. CGF comparison. A total of 74 proteins were identified upregulation and 261 were downregulation in the GF mice compared with the CGF mice (Supplementary Table 3). The functional enrichment analysis showed that the AMP-activated protein kinase (AMPK) signaling pathway was the most significantly associated disrupt pathway (p-value=7.59E-04, Fig. 6D). Several preclinical evidences do support the anti-inflammatory actions of AMPK [31]. In the PPI network analysis, the hub proteins with their directly interacted proteins were shown in Fig. 7A, and ten hub proteins were identified (Fig. 7B), e.g., AQR, LSM3, POLR2B, and ZFP830. Several preclinical evidences do support the anti-inflammatory actions of AMPK [31]. In the PPI network. Interestingly, the AQR and LSM3 are also the important components of spliceosome.
Integrated analysis of the proteomic and microarray data from GF mice
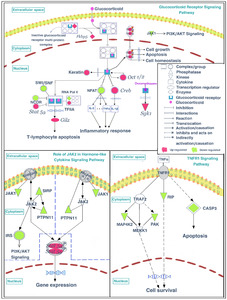
As described in the previous sections, these differentially expressed proteins, especially the upregulated proteins, were primarily enriched in the glucocorticoid receptor signaling pathway, indicating that the absence of gut microbiota may enhance the activity of the glucocorticoid receptor signaling pathway through upregulating the expression of related proteins. To further understand the underlying mechanisms by which gut microbiota regulate functions of glucocorticoid receptor signaling pathway, we performed an integrated analysis of the present proteomic results combined with previously reported microarray data that analyzed changes in the expression of glucocorticoid receptor signaling pathway genes [14]. The dysfunctions of glucocorticoid receptor signaling pathway are associated with disturbed inflammatory response [32]. Stirringly, the present results also indicated the disturbances in inflammation-related pathways in GF mice, that were the role of JAK2 in the hormone-like cytokine signaling pathway and the TNFR1 signaling pathway, and all these inflammation-related proteins were all downregulated in GF mice compared with SPF mice. Finally, the glucocorticoid receptor signaling and inflammation-related pathways were displayed visually in Fig. 8.
DISCUSSION
Gut microbiota plays an important role in regulating the human health and diseases. Increasing evidence proved the potential effects of gut microbiota on mental disorders, e.g., depression and anxiety [33, 34]. However, the underlying mechanisms by which the gut microbiota influence the host’s brain functions and behaviors are complex and remain little understand. Thus, we used iTRAQ-based quantitative proteomic approach to analyze the protein expression changes in the hippocampus of GF, SPF and CGF mice, to further explore the underlying mechanisms at protein level. In present study, inbred line mice were used to mitigate the confounding effects of undefined variation in genetic backgrounds, we found that a total of 303 proteins were dysregulated in GF mice compared with SPF mice, and 124 proteins were significantly restored after gut microbiota colonization. These significant proteins were primarily associated with glucocorticoid receptor signaling and inflammation-related pathways.
As previously reported [35], a complementary approach using CGF mice revealed the restoration of transcript expression, with no normalization of depression- and anxiety-like behavioral changes. Being different from transplanting fecal samples from MDD patients to GF mice that caused significant depressive- and anxiety-like behaviors [15], the CGF mice were naturally colonized with gut microbiota from SPF mice, and this procedure has previously demonstrated to be effective at restoring a normal microbiota [21, 36]. These results suggest that the disturbances in the glucocorticoid receptor signaling and inflammation-related pathways are regulated by gut microbiota, rather than by the depression- or anxiety-like behaviors per se. And previous studies have reported that depression is strongly related to disrupted inflammatory processes [37]. There is growing evidence showed that depression is highly prevalent in infectious and autoimmune diseases, and this co-morbidity cannot be attributed only to the psychological distress of the disease [38]. Moreover, depressed patients consistently have higher levels of pro-inflammatory cytokines, acute phase proteins, chemokines, and cellular adhesion molecules [39, 40]. It has also been reported that an absence of gut microbiota may lead to behavioral abnormalities in mice via the HPA axis, which suggests that gut microbiota are an environmental factor that can regulate the pressure response of the HPA axis. For example, after the same restraint stress, serum adrenocorticotropic hormone levels are significantly higher in GF mice than in SPF mice [22, 41]. Taken together, these results indicate that disturbances in glucocorticoid receptor signaling and inflammation-related pathways in the hippocampus are involved in mechanisms underlying gut microbiota–host interactions in mice.
In the glucocorticoid receptor signaling pathway, Fbpk5, a co-chaperone that participates in the regulation of glucocorticoid receptor sensitivity and the efficiency of the negative feedback process of the HPA axis [42], was upregulated in GF mice compared with SPF mice. FKBP5 and other components form an inactive glucocorticoid receptor (GR) multiprotein complex in the cytoplasm that is released after glucocorticoid combines with GR (GLGR). A previous study consistently found that GF mice had significantly higher levels of Fkbp5 transcripts in the pituitary compared with SPF unstressed mice [43]. It has also been reported that stress upregulates Fkbp5 in the pituitary of male mice compared with unstressed animals [44], although this result may have been caused by the particular stress model that was used. In the present study, RNA polymerase II (RNA pol II), Nuclear receptor corepressor (NCOR), and components of the SWI/SNF complex were downregulated in GF mice compared with SPF mice, and expression levels were restored after gut microbiota colonization. In contrast, Transcription initiation factor IIA (TF IIA) was upregulated in GF mice compared with SPF mice. The transcriptional regulatory complex formed by RNA pol II, TF IIA, and other proteins can interact with GLGR dimers and further influence downstream gene expression. In addition, nuclear factor of activated T cells (NFAT), a transcription factor involved in the transcriptional regulation of inflammatory genes, was downregulated in GF mice compared with SPF mice. Moreover, Stat5a and Creb can regulate the activation of transcription, and were upregulated in GF mice in the previous study [14]. Phosphorylated STAT5a protein in the nucleus promotes cell growth, apoptosis, and cell homeostasis functions. cAMP responsive element binding protein 1 (CREB) can promote the synthesis of IL1-β and is repressed by the GLGR complex, followed by the disruption of the inflammatory response. Studies have found that treatment with Bifidobacterium breve CCFM1025 can reduce depression- and anxiety-like behavior in mice, as well as decrease the active HPA axis and inflammatory response. However, increased IL1-β in the hippocampus of stressed mice was not reduced with this treatment [45]. This result is consistent with our findings of upregulated CREB in the hippocampus of GF mice. Additionally, a previous study has reported that commensal microbiota cause an upregulation in the expression of cytokines, including IL-4, IL-5, IL-6, IL-10, IL-13, IL-17, TNF, and IFN, in the colon [43]. Considering the antidepression- and antianxiety-like behaviors in GF mice compared with SPF mice [14], together with the finding that GLGR dimers can regulate the inflammatory response through gene regulation [46-49], it appears that the inflammatory response may play an important role in the mechanisms by which gut microbiota regulate depression- and anxiety-related behaviors.
In inflammation-related pathways, four proteins were enriched in the role of JAK2 in the hormone-like cytokine signaling pathway, they were Janus kinase 1 (JAK1), signal regulatory protein alpha (SIRP), protein tyrosine phosphatase non-receptor type 11 (PTPN11) and insulin receptor substrate 2 (IRS). Four proteins were enriched in the TNFR1 signaling pathway, they were receptor interacting serine/threonine-protein kinase 1 (RIP), Mitogen-activated protein kinase kinase kinase kinase 2 (MAP4K2), p21 (CDKN1A)-activated kinase 6 (PAK), and Caspase-3 (CASP3). These inflammation-related proteins were all downregulated in GF mice compared with SPF mice. Interestingly, a previous study reported that upregulated cytokine expression in vivo can lead to the increased secretion of steroid hormones by activating the HPA axis [50], thus increasing glucocorticoids in the entire body. Conversely, changes in the activity of the glucocorticoid receptor signaling pathway can lead to corresponding changes in the inflammatory pathway [51]. The present proteomic results revealed that the upregulated proteins in the GF vs. SPF comparison were primarily enriched in the glucocorticoid receptor signaling pathway, while the downregulated proteins were highly involved in inflammation-related pathways. Moreover, previous reports have suggested that anti-inflammatory treatments, such as glucocorticoids, which have been used clinically for decades as potent anti-inflammatory and immunosuppressive agents [52], may produce antidepressant effects [53, 54]. Together, these findings suggest that an enhancement of anti-inflammatory functions and a reduction in inflammatory activity may play a synergistic role in the generation of antidepressive and antianxiety behaviors in GF mice.
Some limitations in the present study need to be acknowledged. First, only hippocampal tissue was analyzed in the present study. Future research thus needs to include multiple brain regions to gain a comprehensive understanding of the underlying mechanisms of microbial–host interactions. In addition, cellular-specificity analysis is warranted to get better insight into the role of each hippocampal cell subtype in the microbiota-directed effects within the brain. Second, only male mice were analyzed for proteomic changes in this study, and further studies including both male and female mice are needed to identify any sex-specific effects of gut microbiota on host brain functions. Third, further researches analyzing the genetic background of mouse are required to evaluate its effects on experimental results.
In conclusion, numerous differentially expressed proteins were identified through a proteomic profiling analysis of hippocampal tissue from GF, SPF, and CGF mice, and these significant proteins were mainly related to the glucocorticoid receptor signaling pathway and inflammation-related pathways revealed by bioinformatics analysis. The present results indicate that these disrupted pathways may be involved in the underlying mechanisms by which gut microbiota regulate brain functions and behavioral phenotypes. This study provides new insights into the pathological mechanisms of gut microbiota-regulated diseases.
ACKNOWLEDGEMENTS
This work was supported by the National Key R&D Program of China (Grant no. 2017YFA0505700), the Non-profit Central Research Institute Fund of Chinese Academy of Medical Sciences (Grant no. 2019PT320002) and the Natural Science Foundation Project of China (Grant no. 81820108015). The authors thank the Experimental Animal Research Center at Third Military Medical University (Chongqing, China) for providing the germ-free mice.
CONFLICT OF INTEREST
The authors report no conflicts of interest in this work.
Figures

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
References
- Qin J, Li R, Raes J, Arumugam M, Burgdorf KS, Manichanh C, Nielsen T, Pons N, Levenez F, Yamada T, Mende DR, Li J, Xu J, Li S, Li D, Cao J, Wang B, Liang H, Zheng H, Xie Y, Tap J, Lepage P, Bertalan M, Batto JM, Hansen T, Le Paslier D, Linneberg A, Nielsen HB, Pelletier E, Renault P, Sicheritz-Ponten T, Turner K, Zhu H, Yu C, Li S, Jian M, Zhou Y, Li Y, Zhang X, Li S, Qin N, Yang H, Wang J, Brunak S, Doré J, Guarner F, Kristiansen K, Pedersen O, Parkhill J, Weissenbach J; MetaHIT Consortium, Bork P, Ehrlich SD, Wang J (2010) A human gut microbial gene catalogue established by metagenomic sequencing. Nature 464:59-65



- Bauer KC, Huus KE, Finlay BB (2016) Microbes and the mind: emerging hallmarks of the gut microbiota-brain axis. Cell Microbiol 18:632-644
- Cryan JF, O'Mahony SM (2011) The microbiome-gut-brain axis: from bowel to behavior. Neurogastroenterol Motil 23:187-192
- Mayer EA (2011) Gut feelings: the emerging biology of gut-brain communication. Nat Rev Neurosci 12:453-466
- Rhee SH, Pothoulakis C, Mayer EA (2009) Principles and clinical implications of the brain-gut-enteric microbiota axis. Nat Rev Gastroenterol Hepatol 6:306-314
- Sherwin E, Sandhu KV, Dinan TG, Cryan JF (2016) May the force be with you: the light and dark sides of the microbiota-gut-brain axis in neuropsychiatry. CNS Drugs 30:1019-1041
- Byrne G, Rosenfeld G, Leung Y, Qian H, Raudzus J, Nunez C, Bressler B (2017) Prevalence of anxiety and depression in patients with inflammatory bowel disease. Can J Gastroenterol Hepatol 2017:6496727
- Panara AJ, Yarur AJ, Rieders B, Proksell S, Deshpande AR, Abreu MT, Sussman DA (2014) The incidence and risk factors for developing depression after being diagnosed with inflammatory bowel disease: a cohort study. Aliment Pharmacol Ther 39:802-810
- Huo R, Zeng B, Zeng L, Cheng K, Li B, Luo Y, Wang H, Zhou C, Fang L, Li W, Niu R, Wei H, Xie P (2017) Microbiota modulate anxiety-like behavior and endocrine abnormalities in hypothalamic-pituitary-adrenal axis. Front Cell Infect Microbiol 7:489
- Chen JJ, Xie J, Zeng BH, Li WW, Bai SJ, Zhou C, Chen W, Wei H, Xie P (2019) Absence of gut microbiota affects lipid metabolism in the prefrontal cortex of mice. Neurol Res 41:1104-1112
- Zhou C, Rao X, Wang H, Zeng B, Yu Y, Chen J, Zhong J, Qi X, Zeng L, Zheng P, Hong W, Xie P (2020) Hippocampus-specific regulation of long non-coding RNA and mRNA expression in germ-free mice. Funct Integr Genomics 20:355-365
- Zeng L, Zeng B, Wang H, Li B, Huo R, Zheng P, Zhang X, Du X, Liu M, Fang Z, Xu X, Zhou C, Chen J, Li W, Guo J, Wei H, Xie P (2016) Microbiota modulates behavior and protein kinase C mediated cAMP response element-binding protein signaling. Sci Rep 6:29998
- Chen JJ, Zheng P, Liu YY, Zhong XG, Wang HY, Guo YJ, Xie P (2018) Sex differences in gut microbiota in patients with major depressive disorder. Neuropsychiatr Dis Treat 14:647-655
- Luo Y, Zeng B, Zeng L, Du X, Li B, Huo R, Liu L, Wang H, Dong M, Pan J, Zheng P, Zhou C, Wei H, Xie P (2018) Gut microbiota regulates mouse behaviors through glucocorticoid receptor pathway genes in the hippocampus. Transl Psychiatry 8:187
- Zheng P, Zeng B, Zhou C, Liu M, Fang Z, Xu X, Zeng L, Chen J, Fan S, Du X, Zhang X, Yang D, Yang Y, Meng H, Li W, Melgiri ND, Licinio J, Wei H, Xie P (2016) Gut microbiome remodeling induces depressive-like behaviors through a pathway mediated by the host's metabolism. Mol Psychiatry 21:786-796
- Chen JJ, Zeng BH, Li WW, Zhou CJ, Fan SH, Cheng K, Zeng L, Zheng P, Fang L, Wei H, Xie P (2017) Effects of gut microbiota on the microRNA and mRNA expression in the hippocampus of mice. Behav Brain Res 322(Pt A):34-41
- Liu L, Wang H, Yu Y, Zeng B, Rao X, Chen J, Zhou C, Zheng P, Pu J, Yang L, Zhang H, Wei H, Xie P (2020) Microbial regulation of a lincRNA-miRNA-mRNA network in the mouse hippocampus. Epigenomics 12:1377-1387
- Campbell S, Macqueen G (2004) The role of the hippocampus in the pathophysiology of major depression. J Psychiatry Neurosci 29:417-426
- Huang C, Yang X, Zeng B, Zeng L, Gong X, Zhou C, Xia J, Lian B, Qin Y, Yang L, Liu L, Xie P (2019) Proteomic analysis of olfactory bulb suggests CACNA1E as a promoter of CREB signaling in microbiota-induced depression. J Proteomics 194:132-147
- He Y, Li W, Tian Y, Chen X, Cheng K, Xu K, Li C, Wang H, Qu C, Wang C, Li P, Chen H, Xie P (2018) iTRAQ-based proteomics suggests LRP6, NPY and NPY2R perturbation in the hippocampus involved in CSDS may induce resilience and susceptibility. Life Sci 211:102-117
- Clarke G, Grenham S, Scully P, Fitzgerald P, Moloney RD, Shanahan F, Dinan TG, Cryan JF (2013) The microbiome-gut-brain axis during early life regulates the hippocampal serotonergic system in a sex-dependent manner. Mol Psychiatry 18:666-673
- Sudo N, Chida Y, Aiba Y, Sonoda J, Oyama N, Yu XN, Kubo C, Koga Y (2004) Postnatal microbial colonization programs the hypothalamic-pituitary-adrenal system for stress response in mice. J Physiol 558(Pt 1):263-275
- Unwin RD, Griffiths JR, Whetton AD (2010) Simultaneous analysis of relative protein expression levels across multiple samples using iTRAQ isobaric tags with 2D nano LC-MS/MS. Nat Protoc 5:1574-1582
- Guo H, Huang ZL, Wang W, Zhang SX, Li J, Cheng K, Xu K, He Y, Gui SW, Li PF, Wang HY, Dong ZF, Xie P (2017) iTRAQ-based proteomics suggests Ephb6 as a potential regulator of the ERK pathway in the prefrontal cortex of chronic social defeat stress model mice. Proteomics Clin Appl 11. doi: 10.1002/prca.20170011.
- Iqbal J, Zhang K, Jin N, Zhao Y, Liu Q, Ni J, Shen L (2018) Effect of sodium selenate on hippocampal proteome of 3×Tg-AD mice-exploring the antioxidant dogma of selenium against Alzheimer's disease. ACS Chem Neurosci 9:1637-1651
- Krämer A, Green J, Pollard J Jr, Tugendreich S (2014) Causal analysis approaches in Ingenuity Pathway Analysis. Bioinformatics 30:523-530
- Szklarczyk D, Franceschini A, Wyder S, Forslund K, Heller D, Huerta-Cepas J, Simonovic M, Roth A, Santos A, Tsafou KP, Kuhn M, Bork P, Jensen LJ, von Mering C (2015) STRING v10: protein-protein interaction networks, integrated over the tree of life. Nucleic Acids Res 43(Database issue):D447-D452
- Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, Ramage D, Amin N, Schwikowski B, Ideker T (2003) Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res 13:2498-2504
- Chin CH, Chen SH, Wu HH, Ho CW, Ko MT, Lin CY (2014) cytoHubba: identifying hub objects and sub-networks from complex interactome. BMC Syst Biol 8 Suppl 4:S11
- Wang H, Liu L, Rao X, Zeng B, Yu Y, Zhou C, Zeng L, Zheng P, Pu J, Xu S, Cheng K, Zhang H, Ji P, Wei H, Xie P (2020) Integrated phosphoproteomic and metabolomic profiling reveals perturbed pathways in the hippocampus of gut microbiota dysbiosis mice. Transl Psychiatry 10:346
- O'Neill LA, Hardie DG (2013) Metabolism of inflammation limited by AMPK and pseudo-starvation. Nature 493:346-355
- Gupta S, Haldar C (2013) Physiological crosstalk between melatonin and glucocorticoid receptor modulates T-cell mediated immune responses in a wild tropical rodent, Funambulus pennanti. J Steroid Biochem Mol Biol 134:23-36
- Simpson CA, Diaz-Arteche C, Eliby D, Schwartz OS, Simmons JG, Cowan CSM (2020) The gut microbiota in anxiety and depression - a systematic review. Clin Psychol Rev 83:101943
- Warner BB (2019) The contribution of the gut microbiome to neurodevelopment and neuropsychiatric disorders. Pediatr Res 85:216-224
- Chen JJ, Zhou CJ, Zheng P, Cheng K, Wang HY, Li J, Zeng L, Xie P (2017) Differential urinary metabolites related with the severity of major depressive disorder. Behav Brain Res 332:280-287
- Hansen CH, Nielsen DS, Kverka M, Zakostelska Z, Klimesova K, Hudcovic T, Tlaskalova-Hogenova H, Hansen AK (2012) Patterns of early gut colonization shape future immune responses of the host. PLoS One 7:e34043
- Kohler O, Krogh J, Mors O, Benros ME (2016) Inflammation in depression and the potential for anti-inflammatory treatment. Curr Neuropharmacol 14:732-742
- Pollak Y, Yirmiya R (2002) Cytokine-induced changes in mood and behaviour: implications for 'depression due to a general medical condition', immunotherapy and antidepressive treatment. Int J Neuropsychopharmacol 5:389-399
- Raison CL, Capuron L, Miller AH (2006) Cytokines sing the blues: inflammation and the pathogenesis of depression. Trends Immunol 27:24-31
- Howren MB, Lamkin DM, Suls J (2009) Associations of depression with C-reactive protein, IL-1, and IL-6: a meta-analysis. Psychosom Med 71:171-186
- Neufeld KM, Kang N, Bienenstock J, Foster JA (2011) Reduced anxiety-like behavior and central neurochemical change in germ-free mice. Neurogastroenterol Motil 23:255-264
- Bekhbat M, Rowson SA, Neigh GN (2017) Checks and balances: the glucocorticoid receptor and NFĸB in good times and bad. Front Neuroendocrinol 46:15-31
- Vodička M, Ergang P, Hrnčíř T, Mikulecká A, Kvapilová P, Vagnerová K, Šestáková B, Fajstová A, Hermanová P, Hudcovic T, Kozáková H, Pácha J (2018) Microbiota affects the expression of genes involved in HPA axis regulation and local metabolism of glucocorticoids in chronic psychosocial stress. Brain Behav Immun 73:615-624
- Vagnerová K, Vodička M, Hermanová P, Ergang P, Šrůtková D, Klusoňová P, Balounová K, Hudcovic T, Pácha J (2019) Interactions between gut microbiota and acute restraint stress in peripheral structures of the hypothalamic-pituitary-adrenal axis and the intestine of male mice. Front Immunol 10:2655
- Tian P, O'Riordan KJ, Lee YK, Wang G, Zhao J, Zhang H, Cryan JF, Chen W (2020) Towards a psychobiotic therapy for depression:
Bifidobacterium breve CCFM1025 reverses chronic stress-induced depressive symptoms and gut microbial abnormalities in mice. Neurobiol Stress 12:100216
- Adcock IM, Caramori G (2001) Cross-talk between pro-inflammatory transcription factors and glucocorticoids. Immunol Cell Biol 79:376-384
- Adcock IM, Ito K, Barnes PJ (2004) Glucocorticoids: effects on gene transcription. Proc Am Thorac Soc 1:247-254
- De Bosscher K, Vanden Berghe W, Haegeman G (2003) The interplay between the glucocorticoid receptor and nuclear factor-kappaB or activator protein-1: molecular mechanisms for gene repression. Endocr Rev 24:488-522
- Mozo L, Suárez A, Gutiérrez C (2004) Glucocorticoids up-regulate constitutive interleukin-10 production by human monocytes. Clin Exp Allergy 34:406-412
- Zunszain PA, Anacker C, Cattaneo A, Carvalho LA, Pariante CM (2011) Glucocorticoids, cytokines and brain abnormalities in depression. Prog Neuropsychopharmacol Biol Psychiatry 35:722-729
- Silverman MN, Sternberg EM (2012) Glucocorticoid regulation of inflammation and its functional correlates: from HPA axis to glucocorticoid receptor dysfunction. Ann N Y Acad Sci 1261:55-63
- Vandewalle J, Luypaert A, De Bosscher K, Libert C (2018) Therapeutic mechanisms of glucocorticoids. Trends Endocrinol Metab 29:42-54
- Müller N, Schwarz MJ, Dehning S, Douhe A, Cerovecki A, Goldstein-Müller B, Spellmann I, Hetzel G, Maino K, Kleindienst N, Möller HJ, Arolt V, Riedel M (2006) The cyclooxygenase-2 inhibitor celecoxib has therapeutic effects in major depression: results of a double-blind, randomized, placebo controlled, add-on pilot study to reboxetine. Mol Psychiatry 11:680-684
- Tyring S, Gottlieb A, Papp K, Gordon K, Leonardi C, Wang A, Lalla D, Woolley M, Jahreis A, Zitnik R, Cella D, Krishnan R (2006) Etanercept and clinical outcomes, fatigue, and depression in psoriasis: double-blind placebo-controlled randomised phase III trial. Lancet 367:29-35