Articles

Article Tools
 View Full Text View Full Text |
 Abstract Abstract |
 Article as PDF Article as PDF |
 Print this Article Print this Article |
 Pubmed Pubmed |
 PMC PMC |
 PubReader PubReader |
 Export to Citation Export to Citation |
 Email Alerts Email Alerts |
 Open Access Open Access |
Share this article on :



Stats or Metrics
Article
Original Article
Exp Neurobiol 2021; 30(5): 329-340
Published online October 31, 2021
https://doi.org/10.5607/en21018
© The Korean Society for Brain and Neural Sciences
Upregulation of Toll-like Receptor 2 in Dental Primary Afferents Following Pulp Injury
Pa Reum Lee†, Jin-Hee Lee†, Ji Min Park and Seog Bae Oh*
Department of Neurobiology and Physiology, School of Dentistry and Dental Research Institute, Seoul National University, Seoul 03080, Korea
Correspondence to: *To whom correspondence should be addressed.
TEL: 82-2-740-8656, FAX: 82-2-762-5107
e-mail: odolbae@snu.ac.kr
†These authors contributed equally to this work.
This is an Open Access article distributed under the terms of the Creative Commons Attribution Non-Commercial License (http://creativecommons.org/licenses/by-nc/4.0) which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
Abstract
Pulpitis (toothache) is a painful inflammation of the dental pulp and is a prevalent problem throughout the world. This pulpal inflammation occurs in the cells inside the dental pulp, which have host defense mechanisms to combat oral microorganisms invading the pulp space of exposed teeth. This innate immunity has been well studied, with a focus on Toll-like receptors (TLRs). The function of TLR4, activated by Gram-negative bacteria, has been demonstrated in trigeminal ganglion (TG) neurons for dental pain. Although Gram-positive bacteria predominate in the teeth of patients with caries and pulpitis, the role of TLR2, which is activated by Gram-positive bacteria, is poorly understood in dental primary afferent (DPA) neurons that densely innervate the dental pulp. Using Fura-2 based Ca2+ imaging, we observed reproducible intracellular Ca2+ responses induced by Pam3CSK4 and Pam2CSK4 (TLR2-specific agonists) in TG neurons of adult wild-type (WT) mice. The response was completely abolished in TLR2 knock-out (KO) mice. Single-cell RT-PCR detected
Graphical Abstract
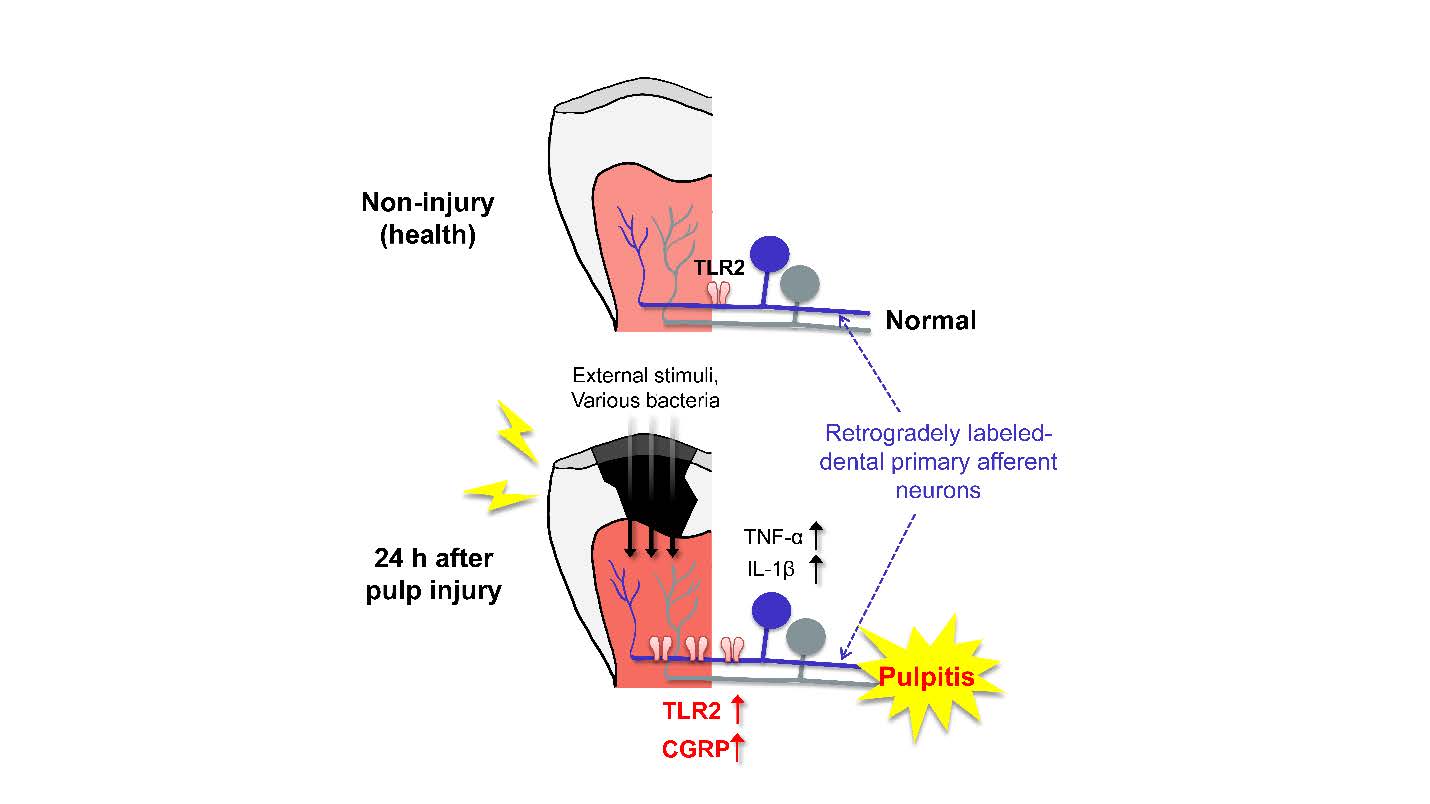
Keywords: Neurobiology, Dental pulp exposure, Pulpitis, Toll-like receptors, Trigeminal nerve
INTRODUCTION
Pulpitis (toothache) is a painful inflammation of the dental pulp and is a problem of high prevalence throughout the world. It is caused primarily by oral bacteria that infect the pulp, where blood vessels and dental primary afferent (DPA) neurons are richly supplied [1]. The most common entry points of these oral bacteria are dental caries, followed by dental trauma or cracked teeth [2]. The major organisms in dental caries are Gram-positive bacteria, such as
Pulpitis commonly accompanies the innate dental pulp immunity that is mediated by the initial sensing of the bacterial pathogens, intracellular signal transduction, and the release of by-products, such as cytokines and inflammatory mediators [1, 2]. A well-known mechanism underlying this innate immunity is the activation of pattern recognition receptors (PRRs) that recognize pathogen-associated molecular patterns (PAMPs) in odontoblasts [1, 10], pulp fibroblasts [11], and sentinel cells [1, 11]. A highly relevant class of PRRs is the family of Toll-like receptors (TLRs), of which 12 TLRs have been found in mammals [12]; therefore, TLRs play an essential role in the first-line defense of the dental pulp.
As the known cell surface TLRs in rodents are TLR1, TLR2, TLR4, TLR5, and TLR6, they can bind and respond to invading bacteria or their specific structural motifs [13, 14]. For example, TLR4 is activated by lipopolysaccharides present in the cell wall of Gram-negative bacteria, whereas TLR2, which forms heterodimers with either TLR1 or TLR6, is activated by peptidoglycan, lipoteichoic acid (LTA), or lipoproteins derived from Gram-positive bacteria [14]. Gram-positive bacteria predominate over Gram-negative bacteria on the teeth of patients diagnosed with caries and/or pulpitis [3, 4], and upregulation of TLR2 is greater than that of TLR4 in the dental pulp exposed to the oral environment [15]. Nevertheless, the contribution of TLR2 expressed in DPA neurons is not yet fully understood, while the role of TLR4 expressed in trigeminal ganglion (TG) neurons has been studied on dental pulp inflammatory pain [16-19].
Thus, in this study, we focused on the functional and molecular expression of TLR2 in DPA neurons and its change in pulpal inflammation from adult mice. We examined whether TLR2 expressed in TG neurons responds to the synthetic bacterial lipopeptides, Pam3CSK4 and Pam2CSK4. Next, we examined
MATERIALS AND METHODS
Animals
A total of 39 male C57BL/6J WT mice (5 to 8-week-old) (Doo Yeol Biotech, South Korea), two male transient receptor potential vanilloid 1 (TRPV1) KO mice, and four male TRPV1-ZsGreen mice (crossed the Trpv1-Cre mice [20] with Ai6 Rosa26ZsGreen1 mice) of C57BL/6J background (5 to 7-week-old) (Jackson Laboratory, USA) were used for this study. A total of eight male and female TLR2 KO mice (5 to 7-week-old) were kindly provided by Dr. Seung Hyun Han (Seoul National University, South Korea). The mice were maintained on ad libitum standard lab chow (pellets) and water under standard conditions (23±1℃, 12 h light-dark cycle). All the surgical and experimental procedures were approved by the Institutional Animal Care and Use Committee (IACUC) of Seoul National University (protocol code: SNU-190313-6). This study also conformed to the ARRIVE (Animal Research: Reporting In Vivo Experiments) guidelines for preclinical animal studies.
Retrograde labeling of DPA neurons with DiI and primary culture of neurons
All the procedures were performed using the method described previously [21]. Briefly, the pulp of the maxillary first molar was exposed with a low-speed dental drill after induction of anesthesia with pentobarbital (60 to 70 mg/kg, intraperitoneal). The cavity was then filled with crystals of DiI (Molecular Probes, USA) and was occluded with a glass ionomer sealant (GC Fuji II, GC Corporation, Japan). The TG neurons containing DiI-labeled DPA neurons were cultured after two weeks post-labeling.
Single-cell reverse transcriptase polymerase chain reaction (scRT-PCR)
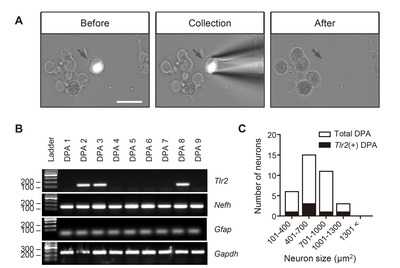
The DPA neurons were visible with DiI labeling, and they were collected by micromanipulation using a glass pipette. Each neuron was photographed for measuring neuron size (µm2) that could represent neuronal types (e.g., small to medium size for nociceptive neurons and large size for non-nociceptive neurons) before cell collection. A single neuron was placed into each tube containing reverse transcription agents (Superscript III, Invitrogen, USA). Complementary DNA (cDNA) synthesis and nested PCR amplification using nested primer pairs (listed in Table 1) were sequentially performed as previously described [21].
Retrograde labeling of DPA neurons with Fluoro-Gold (FG) before dental pulp injury
Following previously described methods [22], the enamel surfaces of the maxillary first molars on both sides were peeled using a low-speed dental drill under pentobarbital anesthesia (60 to 70 mg/kg, intraperitoneal). Then, we performed an etching with a 32% phosphoric acid etchant (Denkist, South Korea) for approximately 10 s, followed by irrigation using a sterilized saline solution. The cavity was filled with crystals of FG (Fluorochrome, USA) and was occluded with a glass ionomer sealant.
Dental pulp injury
One week post-labeling with FG, the pulp of the right maxillary first molar in each mouse was exposed using a low-speed dental drill, as previously described [23]. Dentinal debris was removed from the dental pulp surface using dental k-files (#06, MANI, INC., Japan), followed by irrigation with a sterilized saline solution. After recovering from the anesthetic, the mice were returned to their cages and were supplied with regular food and water.
Real-time RT-PCR
The mice were sacrificed at 24 h post-injury and were perfused with diethyl pyrocarbonate (DEPC) (Sigma-Aldrich, USA)-treated phosphate-buffered saline (PBS) solution (pH 7.4) to prevent blood contaminants. The TG tissues were then harvested immediately and were used for RNA isolation. Total RNA (250 ng), which was purified using an RNeasy Mini Plus Kit (Qiagen, Germany), was synthesized to cDNA using a QuantiTect Reverse Transcription kit (Qiagen, Germany) according to the manufacturer’s instructions. All the PCR amplification procedures were performed as previously described [21], and the relative expressions of the genes were calculated using the ΔΔCT method with
Immunohistochemistry
At 24 h post-injury, the mice were perfused and fixed with 4% paraformaldehyde (PFA) solution, and the harvested TG tissues were transversely sectioned (thickness: 14 µm). The TG sections were incubated with a blocking solution containing 10% normal rabbit serum (NRS) in PBS containing 0.3% Triton X-100 (PBST) and then were stained with rat anti-TLR2 antibody (1:100 dilution, Abcam, Cat. #ab1186, USA) in 0.3% PBST containing 1% NRS. As a secondary antibody, rabbit anti-rat Cy3 (1:200 dilution, Jackson ImmunoResearch, USA) was used with the same diluents of the primary antibody, and NeuroTrace 640/660 Deep-Red Fluorescent Nissl Stain (1:100 dilution, Molecular Probes, USA) was followed to identify the cell bodies and their sizes (data not shown). For double immunostaining for TLR2 and CGRP, we sequentially incubated a second blocking solution containing 10% normal donkey serum (NDS) in 0.3% PBST after TLR2 immunostaining, followed by goat anti-CGRP (1:1,000 dilution, Abcam, Cat. #ab36001, USA) in 0.3% PBST containing 1% NDS and donkey anti-goat Alexa Fluor 647 (1:200 dilution, Jackson ImmunoResearch, USA) in the same diluents of the primary antibody. All images were examined using a confocal microscope (LSM700, Zeiss, Germany).
All the subsequent immunohistochemical analyses, including cross-sectional area (µm2) and fluorescence intensity of FG-labeled DPA neurons within each TG section, were performed using ImageJ (National Institutes of Health, USA). A total of 3 to 6 TG sections per mouse were randomly selected. The image of the selected sections was converted to 8-bit greyscale, and the brightness/contrast and color threshold were adjusted equally for all the images. Each neuron was determined to be either positive or negative based on its mean intensity value.
Ratiometric Ca2+ imaging
TG neurons were used for Ca2+ imaging within 24 h of the primary culture. Fura-2-based ratiometric Ca2+ imaging was performed as previously described [21]. Fura-2 AM (2 μM; Invitrogen, USA) was loaded for one hour at 37℃. A bath solution (containing 140 mM NaCl, 5 mM KCl, 2 mM CaCl2, 1 mM MgCl2, 10 mM HEPES, and 10 mM glucose) and a zero Ca2+ bath solution (containing 140 mM NaCl, 5 mM KCl, 2 mM EGTA, 1 mM MgCl2, 10 mM HEPES, and 10 mM glucose) were adjusted to pH 7.3±0.1 with NaOH and were prepared fresh before each experiment. The 340/380 ratiometric images and [Ca2+]i changes were acquired using MetaFluor software (v 7.8.13.0; Molecular Devices, USA). In all the Ca2+ imaging experiments, TLR2 agonist-positive neurons, displayed as Ca2+ transients with ≥7% of the maximal amplitude (in 50 mM KCl) and reproducibility of at least two out of three, were used for the analysis.
Chemicals
Pam3CSK4 (Invivogen, Cat. #tlrl-pms, USA) and Pam2CSK4 (Invivogen, Cat. #tlrl-pm2s-1, USA) were dissolved in the supplied endotoxin-free water (1 mg/ml). Capsaicin (Sigma-Aldrich, USA) was prepared in ethanol (10 mM) and stored at -20℃. The drugs were diluted to their final concentration with the bath solution for Ca2+ imaging.
Data analysis and statistics
The statistical data are presented as the mean±SEM. The differences between three groups were compared using a one-way ANOVA with a Bonferroni post hoc test, and the differences between two groups were compared using a two-tailed Student’s
RESULTS
Pam3CSK4 induces intracellular Ca2+ ([Ca2+]i) responses in TG neurons
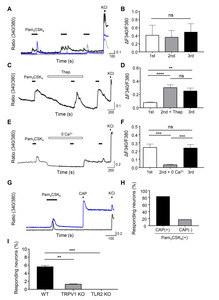
To determine the functional expression patterns of TLR2, we chose to use in our study the synthetic bacterial lipopeptide Pam3CSK4 (a TLR2/TLR1-specific agonist) typically used for TLR2 activation [13, 24]. Activation of TLR2 by three sequential applications of Pam3CSK4 (1 μg/ml; 100 s/application) elicited reproducible [Ca2+]i responses in a subset of TG neurons (Fig. 1A, 1B). When we further confirmed the source of the Ca2+, the Pam3CSK4-induced [Ca2+]i responses were increased even by depletion of the [Ca2+]i store with a 1 μM thapsigargin pretreatment, but the Ca2+ response remained unchanged by the following Pam3CSK4 application after washout with bath solution (Fig. 1C, 1D). Conversely, the [Ca2+]i responses were completely abolished by a zero Ca2+ bath solution (Fig. 1E, 1F) and in TLR2 KO mice (Fig. 1I). Among the 50 mM KCl-responsive TG neurons, a subset of TG neurons responded to Pam3CSK4 (approximately 5.7%,
Pam2CSK4 induces [Ca2+]i responses in TG neurons
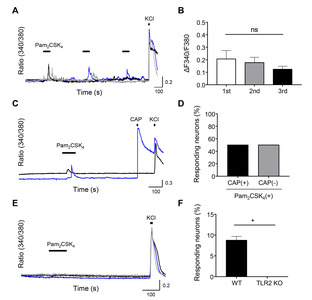
Similar to Pam3CSK4, three sequential applications of Pam2CSK4 (a TLR2/TLR6-specific agonist) (1 μg/ml; 100 s/application) elicited reproducible [Ca2+]i responses in a subset of TG neurons (Fig. 2A, 2B). The Pam2CSK4-responding neurons were also observed in TRPV1-positive neurons as well as TRPV1-negative neurons (Fig. 2C), and the CAP (1 µM)-induced Ca2+ transients were elicited in half of the Pam2CSK4-responding neurons (50%,
TLR2 is distinctly expressed in DPA neurons
We investigated the TLR2 expression patterns in individual DPA neurons that were retrogradely labeled with DiI using scRT-PCR (Fig. 3A). Approximately 17.1% of the DPA neurons expressed
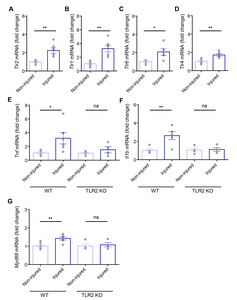
Dental pulp injury promotes TLR2 mRNA expression and pulpal inflammation
Since significant morphological changes and pain-like behaviors can occur within 24 h of the experimental dental pulp injury [17, 23], we assessed the molecular changes to
Expression of TLR2 in DPA neurons is upregulated by pulpal inflammation
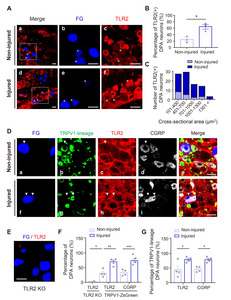
Since non-neuronal cells like infiltrated macrophages can be contained in the injured or inflamed TG tissues [29], we examined the changes that occur in TLR2 protein expression by pulpal inflammation in the cell bodies of DPA neurons that were labeled with the retrograde neuronal tracer FG. Immunohistochemical analysis showed a significant increase of the TLR2-positive DPA neurons in the injured side, compared to the non-injured side, at 24 h post-injury (Fig. 5A, 5B), and the increase in small- to medium-sized neurons (up to 700 µm2) was greater than in large-sized neurons, suggesting that TLR2 has functional roles in nociceptive neurons associated with pain (Fig. 5C). Next, we found that both TLR2 and CGRP (pain-related neuropeptides) expression levels were upregulated in the injured DPA neurons, compared to the non-injured DPA neurons, at 24 h post-injury (Fig. 5D, 5F), and the increase was also observed in TRPV1-lineage DPA neurons, suggesting that upregulation of TLR2 in DPA neurons can contribute to pain during pulpitis (Fig. 5D, 5G). The anti-TLR2 antibody specificity used in our work was verified by a significant decrease in TLR2 immunoreactivity in TLR2 KO mice (Fig. 5E, 5F).
DISCUSSION
In this study, we present a possible role of neuronal TLR2 in pulpitis with the following findings: (1) Both Pam3CSK4 and Pam2CSK4 (TLR2-specific agonists) increased the [Ca2+]i levels in a subset of TG neurons, which were responsive to capsaicin (a TRPV1 agonist), but this response was abolished entirely in TLR2 KO mice. (2)
TLR2 activation triggers downstream signal transduction pathways, usually in non-neuronal cells such as macrophages and epithelial cells, by inducing the release of Ca2+ from intracellular stores [30]. However, the mechanisms for this action are different in sensory neurons such as DRG neurons [24]. Wang et al. [24] has demonstrated that Pam3CSK4 dramatically increases the [Ca2+]i concentrations, depending on the activation of downstream TRPV1 and TRPA1 channels, by confirming a significant inhibition of this response in TRPV1 KO and TRPA1 KO mice. Our Ca2+ imaging results confirmed that Pam3CSK4-induced Ca2+ influxes were obtained from the extracellular source (Fig. 1E, 1F), but not from the intracellular store (Fig. 1C, 1D). Moreover, TRPV1-expressing TG neurons responded to either Pam3CSK4 (approximately 82.6%,
Although we primarily used Pam3CSK4 to verify the function of TLR2 in sensory neurons, the heterodimeric TLR2 receptors (TLR2/TLR1 or TLR2/TLR6) are distinctly activated by different agonists [24]. The Pam3CSK4 is the triacylated lipopeptide commonly derived from Gram-negative bacteria, while Pam2CSK4 is the diacylated lipopeptide derived from Gram-positive bacteria [31]. Compared to Pam3CSK4 actions (approximately 5.7%,
The experimental dental pulp injury model used in this study is well established and reviewed for studying neuroinflammatory interactions by allowing the molecular changes in the pulp tissue and somatic TG induced by dental injury [17, 23, 37, 38]. Using this model, we confirmed that 24 h following dental pulp injury is a sufficient amount of time to address the upregulation of (1) TLR2 expression in both the injured TG (Fig. 4A) and in (2) individual DPA neurons on the injured side (Fig. 5B) and especially (3) inflammatory marker genes, such as TNF-α and IL-1β (Fig. 4E, 4F), and MYD88, which is associated with the TLR downstream signaling pathway (Fig. 4G), in injured TG from WT mice, but not from TLR2 KO mice. Although this model cannot completely exclude the damage itself following pulpal injury, it sufficiently allows for bacterial accumulation in the exposed dental cavity [37, 39]. Accordingly, our study has limitations in demonstrating that molecular changes are caused by PAMPs but not by damage-associated molecular patterns (DAMPs). However, we showed a significant increase of both TLR2 and CGRP expression levels in TRPV1-lineage DPA neurons, which have been reported to be upregulated by bacterial infection [40], suggesting that upregulation of TLR2 can contribute to pain transmission during pulpitis (Fig. 5F, 5G).
Mammals host a complex community of commensal bacteria, with an estimated 200 to 700 species residing in the oral cavity [5, 41]. Dental caries is commonly caused by Gram-positive bacteria, such as
In conclusion, our results provide a better molecular understanding of pulpitis by revealing that TLR2 expression is upregulated in DPA neurons, and this study uncovers potential neural functions of TLR2 in pulpal inflammation.
ACKNOWLEDGEMENTS
Authors thank Dr. Seung Hyun Han (School of Dentistry, Seoul National University) for providing TLR2 KO mice. This research was supported by National Research Foundation (NRF) of Korea (grant number: 2017M3C7A1025602, 2018R1A5A2024418, and 2021R1A2C3003334) funded by the Korean government (Ministry of Education, Science and Technology). All authors gave their final approval and agree to be accountable for all aspects of the work.
CONFLICT OF INTEREST
All authors declare no conflict of interest.
Figures
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Tables
List of primers used
| scRT-PCR | |||
| Target gene | Outer primer pairs | Inner primer pairs | GenBank no. |
| (F) 5’-GTTCCTACCCCCAATGTGTCC-3’ | (F) 5’-TTGTGATGGGTGTGAACCAC-3’ | NM_001289726.1, NM_008084.3 | |
| (R) 5’-ACCTGTTGCTGTAGCCGTATC-3’ | (R) 5’-CCTTCCACAATGCCAAAGTTG-3’ | ||
| (F) 5’-TGCGTATAGACAGGAGGCAGA-3’ | (F) 5’-TGGAGGAGGAGATCCAGTTCTT-3’ | NM_001131020.1, NM_010277.3 | |
| (R) 5’-GGCGATAGTCGTTAGCTTCGT-3’ | (R) 5’-GCCACTGCCTCGTATTGAGT-3’ | ||
| (F) 5’-GCAGCCAAAGTGAACACAGA-3’ | (F) 5’-AGAGTTGGAGGCCCTGAAAA-3’ | NM_010904.3 | |
| (R) 5’-AATGTCCAGGGCCATCTTGA-3’ | (R) 5’-GTGCAGCCATCTCCCACTT-3’ | ||
| (F) 5’-TCTCTGGGCAGTCTTGAACA-3’ | (F) 5’-AGACACTGGGGGTAACATCG-3’ | NM_011905.3 | |
| (R) 5’-TCGCGGATCGACTTTAGACT-3’ | (R) 5’-AGAGAAGTCAGCCCAGCAAA-3’ | ||
| Real-time RT-PCR | |||
| Target gene | Primer pairs | GenBank no. | |
| (F) 5’-CCAGAACATCATCCCTGCAT-3’ (R) 5’-GCATCGAAGGTGGAAGAGTG-3’ | NM_001289726.1, NM_008084.3 | ||
| (F) 5’-AAGGAATGTGACTTCCAGACCA-3’ (R) 5’-AAGTCCTTCTTCATCGCCTTGT-3’ | NM_ 010851.3 | ||
| (F) 5’-GGCTGCTTCCAAACCTTTGAC-3’ (R) 5’-AGCTTCTCCACAGCCACAAT-3’ | NM_008361.4 | ||
| (F) 5’-TCAAGTGTGCAGCTGATTGCT-3’ (R) 5’-TGCTAACGTGCCGAAGAGATC-3’ | NM_001276445.1, NM_030682.2 | ||
| (F) 5’-CCTCTATTCCCTCCGGACTC-3’ (R) 5’-AACCTGGAGGTTCGCACA-3’ | NM_011905.3 | ||
| (F) 5’- AGTGGCCCTACCAAGTCTCA-3’ (R) 5’- GTGTCTCAGGCTGTTTGTTCC-3’ | NM_021297.3 | ||
| (F) 5’-TGAGCCAAGACAGAAAACCCA-3’ (R) 5’-GGGACATGAGTAAGGTTCCTGTT-3’ | NM_001384171.1, NM_011604.5, NM_001359180.2 | ||
| (F) 5’-AGGTTCTCTTCAAGGGACAAGG-3’ (R) 5’-GAGAGGAGGTTGACTTTCTCCTG-3’ | NM_013693.3, NM_001278601.1 | ||
References
- Farges JC, Alliot-Licht B, Renard E, Ducret M, Gaudin A, Smith AJ, Cooper PR (2015) Dental pulp defence and repair mechanisms in dental caries. Mediators Inflamm 2015:230251



- Rechenberg DK, Galicia JC, Peters OA (2016) Biological markers for pulpal inflammation: a systematic review. PLoS One 11:e0167289
- Love RM, Jenkinson HF (2002) Invasion of dentinal tubules by oral bacteria. Crit Rev Oral Biol Med 13:171-183
- Mallya PS, Mallya S (2020) Microbiology and clinical implications of dental caries - a review. J Evol Med Dent Sci 9:3670-3675
- Loesche WJ (1996) Microbiology of dental decay and periodontal disease. In: Medical microbiology (Baron S ed). 4th ed. Chapter 99. University of Texas Medical Branch at Galveston, Galveston (TX)
- Popova C, Dosseva-Panova V, Panov V (2013) Microbiology of periodontal diseases. A review. Biotechnol Biotechnol Equip 27:3754-3759
- Gao L, Xu T, Huang G, Jiang S, Gu Y, Chen F (2018) Oral microbiomes: more and more importance in oral cavity and whole body. Protein Cell 9:488-500
- Simón-Soro A, Guillen-Navarro M, Mira A (2014) Metatranscriptomics reveals overall active bacterial composition in caries lesions. J Oral Microbiol 6:25443
- Zarco MF, Vess TJ, Ginsburg GS (2012) The oral microbiome in health and disease and the potential impact on personalized dental medicine. Oral Dis 18:109-120
- Yumoto H, Hirao K, Hosokawa Y, Kuramoto H, Takegawa D, Nakanishi T, Matsuo T (2018) The roles of odontoblasts in dental pulp innate immunity. Jpn Dent Sci Rev 54:105-117
- Staquet MJ, Carrouel F, Keller JF, Baudouin C, Msika P, Bleicher F, Kufer TA, Farges JC (2011) Pattern-recognition receptors in pulp defense. Adv Dent Res 23:296-301
- Janeway CA Jr, Medzhitov R (2002) Innate immune recognition. Annu Rev Immunol 20:197-216
- Ignacio BJ, Albin TJ, Esser-Kahn AP, Verdoes M (2018) Toll-like receptor agonist conjugation: a chemical perspective. Bioconjug Chem 29:587-603
- Jang JH, Shin HW, Lee JM, Lee HW, Kim EC, Park SH (2015) An overview of pathogen recognition receptors for innate immunity in dental pulp. Mediators Inflamm 2015:794143
- Mutoh N, Tani-Ishii N, Tsukinoki K, Chieda K, Watanabe K (2007) Expression of toll-like receptor 2 and 4 in dental pulp. J Endod 33:1183-1186
- Filippini HF, Molska GR, Zanjir M, Arudchelvan Y, Gong SG, Campos MM, Avivi-Arber L, Sessle BJ (2020) Toll-like receptor 4 in the rat caudal medulla mediates tooth pulp inflammatory pain. Front Neurosci 14:643
- Lin JJ, Du Y, Cai WK, Kuang R, Chang T, Zhang Z, Yang YX, Sun C, Li ZY, Kuang F (2015) Toll-like receptor 4 signaling in neurons of trigeminal ganglion contributes to nociception induced by acute pulpitis in rats. Sci Rep 5:12549
- Ohara K, Shimizu K, Matsuura S, Ogiso B, Omagari D, Asano M, Tsuboi Y, Shinoda M, Iwata K (2013) Toll-like receptor 4 signaling in trigeminal ganglion neurons contributes tongue-referred pain associated with tooth pulp inflammation. J Neuroinflammation 10:139
- Wadachi R, Hargreaves KM (2006) Trigeminal nociceptors express TLR-4 and CD14: a mechanism for pain due to infection. J Dent Res 85:49-53
- Goswami SC, Mishra SK, Maric D, Kaszas K, Gonnella GL, Clokie SJ, Kominsky HD, Gross JR, Keller JM, Mannes AJ, Hoon MA, Iadarola MJ (2014) Molecular signatures of mouse TRPV1-lineage neurons revealed by RNA-Seq transcriptome analysis. J Pain 15:1338-1359
- Lee PR, Lee JY, Kim HB, Lee JH, Oh SB (2020) TRPM8 mediates hyperosmotic stimuli-induced nociception in dental afferents. J Dent Res 99:107-114
- Gibbs JL, Melnyk JL, Basbaum AI (2011) Differential TRPV1 and TRPV2 channel expression in dental pulp. J Dent Res 90:765-770
- Rossi HL, See LP, Foster W, Pitake S, Gibbs J, Schmidt B, Mitchell CH, Abdus-Saboor I (2020) Evoked and spontaneous pain assessment during tooth pulp injury. Sci Rep 10:2759
- Wang TT, Xu XY, Lin W, Hu DD, Shi W, Jia X, Wang H, Song NJ, Zhang YQ, Zhang L (2020) Activation of different heterodimers of TLR2 distinctly mediates pain and itch. Neuroscience 429:245-255
- Fried K, Sessle BJ, Devor M (2011) The paradox of pain from tooth pulp: low-threshold "algoneurons"?. Pain 152:2685-2689
- Won J, Vang H, Lee PR, Kim YH, Kim HW, Kang Y, Oh SB (2017) Piezo2 expression in mechanosensitive dental primary afferent neurons. J Dent Res 96:931-937
- Vang H, Chung G, Kim HY, Park SB, Jung SJ, Kim JS, Oh SB (2012) Neurochemical properties of dental primary afferent neurons. Exp Neurobiol 21:68-74
- Liu XJ, Liu T, Chen G, Wang B, Yu XL, Yin C, Ji RR (2016) TLR signaling adaptor protein MyD88 in primary sensory neurons contributes to persistent inflammatory and neuropathic pain and neuroinflammation. Sci Rep 6:28188
- Shinoda M, Kubo A, Hayashi Y, Iwata K (2019) Peripheral and central mechanisms of persistent orofacial pain. Front Neurosci 13:1227
- Chun J, Prince A (2006) Activation of Ca2+-dependent signaling by TLR2. J Immunol 177:1330-1337
- Schenk M, Belisle JT, Modlin RL (2009) TLR2 looks at lipoproteins. Immunity 31:847-849
- Buddelmeijer N (2015) The molecular mechanism of bacterial lipoprotein modification--how, when and why?. FEMS Microbiol Rev 39:246-261
- Kovacs-Simon A, Titball RW, Michell SL (2011) Lipoproteins of bacterial pathogens. Infect Immun 79:548-561
- Nguyen MT, Matsuo M, Niemann S, Herrmann M, Götz F (2020) Lipoproteins in gram-positive bacteria: abundance, function, fitness. Front Microbiol 11:582582
- Eguchi J, Ishihara K, Watanabe A, Fukumoto Y, Okuda K (2003) PCR method is essential for detecting Mycobacterium tuberculosis in oral cavity samples. Oral Microbiol Immunol 18:156-159
- Wang H, Ren D (2017) Controlling Streptococcus mutans and Staphylococcus aureus biofilms with direct current and chlorhexidine. AMB Express 7:204
- Byers MR, Närhi MV (1999) Dental injury models: experimental tools for understanding neuroinflammatory interactions and polymodal nociceptor functions. Crit Rev Oral Biol Med 10:4-39
- Yamasaki M, Kumazawa M, Kohsaka T, Nakamura H, Kameyama Y (1994) Pulpal and periapical tissue reactions after experimental pulpal exposure in rats. J Endod 20:13-17
- He Y, Gan Y, Lu J, Feng Q, Wang H, Guan H, Jiang Q (2017) Pulpal tissue inflammatory reactions after experimental pulpal exposure in mice. J Endod 43:90-95
- Chung MK, Lee J, Duraes G, Ro JY (2011) Lipopolysaccharide-induced pulpitis up-regulates TRPV1 in trigeminal ganglia. J Dent Res 90:1103-1107
- Deo PN, Deshmukh R (2019) Oral microbiome: unveiling the fundamentals. J Oral Maxillofac Pathol 23:122-128
- Chiu IM (2018) Infection, pain, and itch. Neurosci Bull 34:109-119
- Chiu IM, Pinho-Ribeiro FA, Woolf CJ (2016) Pain and infection: pathogen detection by nociceptors. Pain 157:1192-1193
- Pinho-Ribeiro FA, Verri WA Jr, Chiu IM (2017) Nociceptor sensory neuron-immune interactions in pain and inflammation. Trends Immunol 38:5-19