Articles

Article Tools
 View Full Text View Full Text |
 Abstract Abstract |
 Article as PDF Article as PDF |
 Print this Article Print this Article |
 Pubmed Pubmed |
 PMC PMC |
 PubReader PubReader |
 Export to Citation Export to Citation |
 Email Alerts Email Alerts |
 Open Access Open Access |
Share this article on :



Stats or Metrics
Article
Review Article
Exp Neurobiol 2022; 31(2): 65-88
Published online April 30, 2022
https://doi.org/10.5607/en22004
© The Korean Society for Brain and Neural Sciences
Physiological Roles of Monomeric Amyloid-β and Implications for Alzheimer’s Disease Therapeutics
Hyomin Jeong1,2,3†, Heewon Shin2,3†, Seungpyo Hong2,4,5,6 and YoungSoo Kim1,2,3,4*
1Division of Integrated Science and Engineering, Underwood International College, Yonsei University, Incheon 21983, 2Department of Pharmacy and 3Yonsei Institute of Pharmaceutical Sciences, College of Pharmacy, Yonsei University, Incheon 21983, 4Yonsei Frontier Lab, Yonsei University, Seoul 03722, Korea, 5Division of Pharmaceutical Sciences, School of Pharmacy, University of Wisconsin-Madison, Madison, 6Wisconsin Center for NanoBioSystems, University of Wisconsin-Madison, Madison, WI 53705, USA
Correspondence to: *To whom correspondence should be addressed.
TEL: 82-32-749-4523, FAX: 82-32-749-4105
e-mail: y.kim@yonsei.ac.kr
†These authors contributed equally to this article.
This is an Open Access article distributed under the terms of the Creative Commons Attribution Non-Commercial License (http://creativecommons.org/licenses/by-nc/4.0) which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
Abstract
- Go to
- Abstract
- Graphical Abstract
- INTRODUCTION
- PROTECTION AGAINST MICROBIAL INFECTIONS
- REGULATION OF SYNAPTIC FUNCTIONS
- PROMOTION OF RECOVERY FROM BRAIN INJURY
- A SEALANT FOR THE BLOOD-BRAIN BARRIER AND MODULATION OF ANGIOGENESIS
- INHIBITION OF PROLIFERATING CANCER CELLS
- Aβ-DIRECTED THERAPEUTICS IN CLINICAL TRIALS AND IMPLICATIONS
- ACKNOWLEDGEMENTS
- Figure
- Table
- Reference
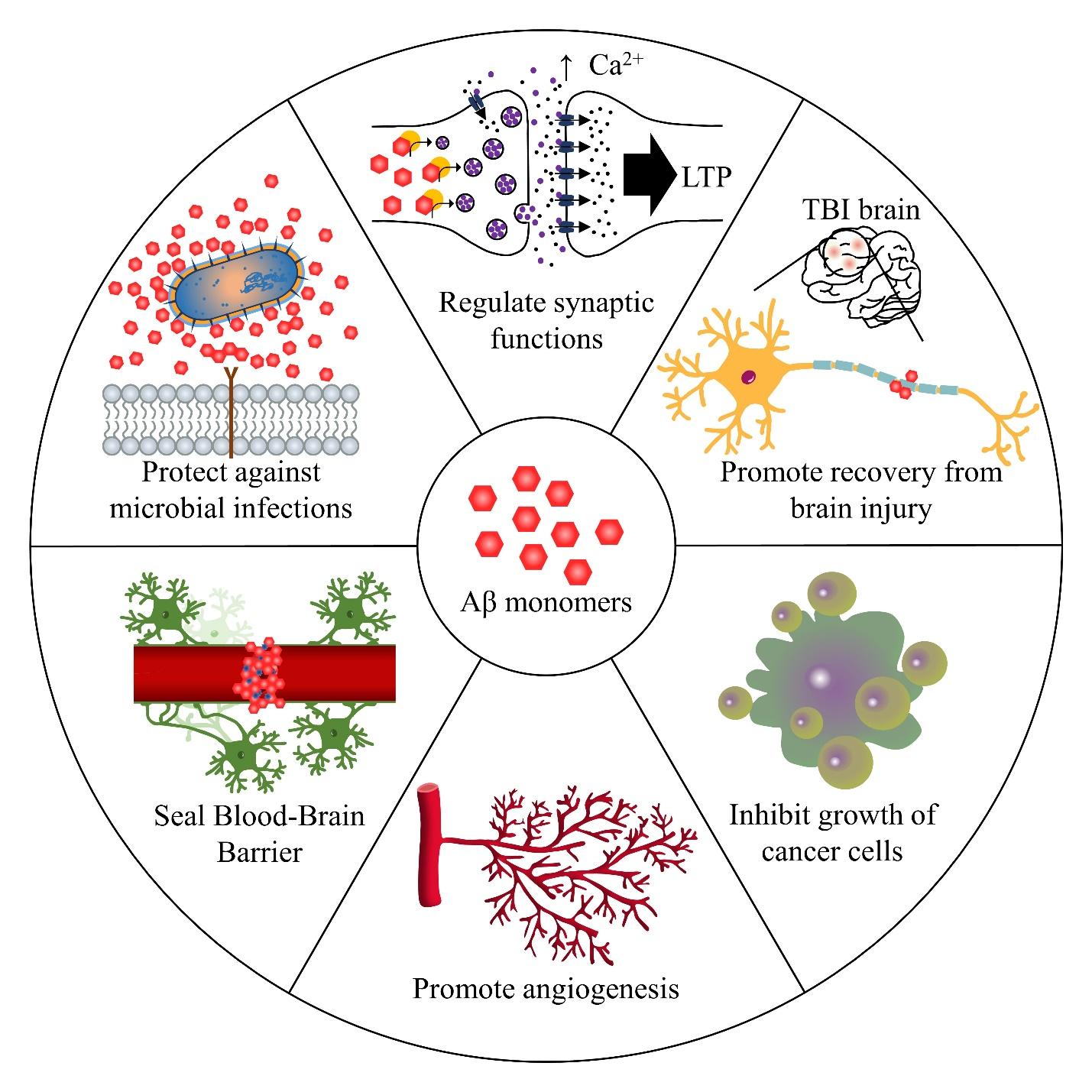
Alzheimer’s disease (AD) progressively inflicts impairment of synaptic functions with notable deposition of amyloid-β (Aβ) as senile plaques within the extracellular space of the brain. Accordingly, therapeutic directions for AD have focused on clearing Aβ plaques or preventing amyloidogenesis based on the amyloid cascade hypothesis. However, the emerging evidence suggests that Aβ serves biological roles, which include suppressing microbial infections, regulating synaptic plasticity, promoting recovery after brain injury, sealing leaks in the blood-brain barrier, and possibly inhibiting the proliferation of cancer cells. More importantly, these functions were found in
Graphical Abstract
- Go to
- Abstract
- Graphical Abstract
- INTRODUCTION
- PROTECTION AGAINST MICROBIAL INFECTIONS
- REGULATION OF SYNAPTIC FUNCTIONS
- PROMOTION OF RECOVERY FROM BRAIN INJURY
- A SEALANT FOR THE BLOOD-BRAIN BARRIER AND MODULATION OF ANGIOGENESIS
- INHIBITION OF PROLIFERATING CANCER CELLS
- Aβ-DIRECTED THERAPEUTICS IN CLINICAL TRIALS AND IMPLICATIONS
- ACKNOWLEDGEMENTS
- Figure
- Table
- Reference
Keywords: Alzheimer’s disease, Amyloid beta-peptides, Amyloidosis, Neurodegenerative diseases, Therapeutics
INTRODUCTION
- Go to
- Abstract
- Graphical Abstract
- INTRODUCTION
- PROTECTION AGAINST MICROBIAL INFECTIONS
- REGULATION OF SYNAPTIC FUNCTIONS
- PROMOTION OF RECOVERY FROM BRAIN INJURY
- A SEALANT FOR THE BLOOD-BRAIN BARRIER AND MODULATION OF ANGIOGENESIS
- INHIBITION OF PROLIFERATING CANCER CELLS
- Aβ-DIRECTED THERAPEUTICS IN CLINICAL TRIALS AND IMPLICATIONS
- ACKNOWLEDGEMENTS
- Figure
- Table
- Reference
Marked by progressive memory loss and cognitive decline, Alzheimer's disease (AD) is the most common neurodegenerative disease that continues to afflict patients and their families without a concrete understanding of its pathogenesis nor effective therapeutics to modify the disease [1-3]. So far, mounting evidence points to the dysregulated accumulation of amyloid-β (Aβ) and tau in the brain as both the cause and biomarker of developing AD. Aβ peptide is known to be derived from amyloid-β precursor protein (AβPP), which can undergo an amyloidogenic or a non-amyloidogenic pathway. When APP is normally cleaved by α-secretase, it goes through the non-amyloidogenic pathway and produces sAPPα and C83 fragment, which are harmless. On the other hand, the dysfunctional amyloidogenic pathway begins by the cleavage of APP by β-secretase and γ-secretase. Here, the β-secretase cleavage yields C99 fragments, and these fragments are further processed into Aβ peptide, which consists of 37 to 49 amino acid residues, depending on the site of γ-secretase cleavage. Aβ peptide mainly exists with 40 amino acid residues, Aβ40, or 42 amino acid residues, Aβ42 [4], which is less abundant, but more pathologically detrimental [5]. In turn, the amyloid cascade follows as Aβ peptides are released into the extracellular space and aggregated into soluble oligomers, fibrils, and plaques, known to damage synapses and neurons. Accordingly, the ‘amyloid cascade hypothesis’ has been postulated arguing that this abnormal deposition of Aβ oligomers and plaques alters the homeostasis of ions and causes synaptic dysfunction, marking Aβ as the primary cause of AD [6-10].
Based on this hypothesis, numerous attempts have been made to alter the course of AD pathogenesis with various means of therapeutics targeting Aβ. However, constant failures in the drug candidates aiming a reduction or complete elimination of Aβ plaques have been reported [11, 12]. For example, reduced levels of Aβ in the brain were observed in phase II trials of verubecestat, a β-secretase inhibitor drug, and solanezumab, a monoclonal antibody drug of Aβ peptide [12, 13], but they showed no efficacy in phase III trials with worsened cognition in some cases [14, 15]. Accordingly, there is an emerging suspicion that questions the conventional approaches of targeting Aβ, or even challenging the amyloid cascade hypothesis to some extent.
Thus, we would like to turn the attention to monomeric form of Aβ. In the past, the monomeric form of Aβ peptide had been widely recognized as a functionless protein generated by APP catabolism, although an evolutionary evidence suggests otherwise: the sequence of Aβ peptide in human has been conserved for at least 400 million years, pointing to the possibility that Aβ perhaps has unidentified, significant roles that confer an evolutionary advantage [16]. Accordingly, we have gathered a significant number of evidence suggesting non-pathological aspects of Aβ monomers and their potential physiological functions. A thorough review of manuscripts has revealed that Aβ may possess antimicrobial properties and play a role in regulating synaptic functions and memory. Also, Aβ may aid the recovery from brain injury with its interference with angiogenesis and even may exert an anticancer effect. Hence, the appreciation of the pathogenesis of AD can be further enhanced by identifying the physiological roles of Aβ monomers, and whether or not to keep Aβ monomers in brains can be determined accordingly to adjust the target of current AD therapeutics.
PROTECTION AGAINST MICROBIAL INFECTIONS
- Go to
- Abstract
- Graphical Abstract
- INTRODUCTION
- PROTECTION AGAINST MICROBIAL INFECTIONS
- REGULATION OF SYNAPTIC FUNCTIONS
- PROMOTION OF RECOVERY FROM BRAIN INJURY
- A SEALANT FOR THE BLOOD-BRAIN BARRIER AND MODULATION OF ANGIOGENESIS
- INHIBITION OF PROLIFERATING CANCER CELLS
- Aβ-DIRECTED THERAPEUTICS IN CLINICAL TRIALS AND IMPLICATIONS
- ACKNOWLEDGEMENTS
- Figure
- Table
- Reference
Aβ and microbes are seemingly unrelated, as the former causes neurodegeneration while infections by the latter result in headache, nausea, and so on; however, antimicrobial activities of Aβ have been postulated by Robinson and Bishop [17] with the ‘Bioflocculant Hypothesis’. The authors have proposed that Aβ binds to neurotoxins and pathogens, and it aggregates into plaques in order to lead the trapped microbes to phagocytes. Later, Moir et al. [18] extended the previous hypothesis by introducing the ‘Antimicrobial Protection Hypothesis’, which notes that Aβ functions as a part of innate immune system, but a certain dysregulation and dysfunction of this pathway lead to an increased Aβ deposition and eventually into a sustained neuroinflammation and neurodegeneration typical of AD patients. This renewed hypothesis not only captures the essence of the antimicrobial function of Aβ, but also provides a plausible explanation for Aβ aggregation.
In supporting these hypotheses, researchers often highlight structural and functional similarities between Aβ and antimicrobial peptides (AMPs), which target specific bacteria, viruses, fungi, and so on to modulate the innate immune system [19]. For instance, Pastore et al. [20] reviewed similarities in sequences and structures between Aβ40 and linear AMPs with highly helical conformations, and Lee et al. [21] summarized their similar propensity to self-assemble into supramolecular structures when faced with bacterial infections. Especially, LL-37, an archetypical human AMP [22], has been frequently compared with Aβ in different studies and review articles to establish the idea that Aβ acts as an AMP [18, 21, 23-27].
In addition to structural comparisons,
Not only the antibacterial and antifungal activities, but also antiviral activities of Aβ have also been reported in a series of
However, compared to the results produced by the synthetic Aβ peptides, those of the cell-derived Aβ exhibited a greater level of adhesion inhibition and agglutination [24]. The answer to this difference may lie on the conformation of Aβ; while the Aβ oligomers are typically removed from the synthetic Aβ peptides, leaving the monomeric form only, the oligomeric form is retained in the cell-derived Aβ. Nonetheless, given that different isoforms of Aβ in physiologically normal human brain have been discovered [31],
Notably, regardless of methods used to prepare Aβ peptides, synthetic or cell-derived, Aβ42 isoform exhibited a greater antibiotic activity than Aβ40 [23, 24]. Accordingly, specific domains of Aβ responsible for the antimicrobial activities were identified as Iso41 and Ala42, which are C-terminal amino acids of Aβ42. The capability of these domains were evidenced by truncated peptides, such as Aβ22-42 and Aβ35-42, causing neutralization and aggregation of influenza A virus and
However, conflicting results were published in regard to the effect of different Aβ isoforms. In contrast to the results provided by Soscia et al. [23] that both Aβ40 and Aβ42 showed an antimicrobial activity against
In line with the
Besides the studies that investigated the direct interaction between pathogens and Aβ, adverse effects reported in clinical trials of β-secretase and γ-secretase inhibitors such as tarenflurbil, semagacestat, and elenbecestat (reviewed by Iqbal et al. [35]) also indirectly signify the potential ability of Aβ against microbial infections. Instead of exhibiting intended clinical benefits such as improved cognitive abilities, the rates of infection elevated in the subjects. Another study demonstrated the regulative effect of different β-secretase and γ-secretase inhibitors, GL-189 and N-[N-(3,5-Difluorophenacetyl)-L-alanyl]-S-phenylglycine-t-butyl-ester (DAPT), on secretion levels of tumor necrosis factor α, interleukin (IL) 6 and 10, which are involved in immune response by macrophages [36]. Intriguingly, these results are in agreement with another
Using the aforementioned
REGULATION OF SYNAPTIC FUNCTIONS
- Go to
- Abstract
- Graphical Abstract
- INTRODUCTION
- PROTECTION AGAINST MICROBIAL INFECTIONS
- REGULATION OF SYNAPTIC FUNCTIONS
- PROMOTION OF RECOVERY FROM BRAIN INJURY
- A SEALANT FOR THE BLOOD-BRAIN BARRIER AND MODULATION OF ANGIOGENESIS
- INHIBITION OF PROLIFERATING CANCER CELLS
- Aβ-DIRECTED THERAPEUTICS IN CLINICAL TRIALS AND IMPLICATIONS
- ACKNOWLEDGEMENTS
- Figure
- Table
- Reference
Contrary to the common notion that Aβ peptides, especially the soluble oligomeric forms, are responsible for impairing synaptic functions followed by disrupted long-term potentiation (LTP) and synaptic plasticity typically seen in AD patients [8-10], increasing evidence suggests that Aβ may play an essential role in synaptic plasticity and memory when maintained at a normal level [42-46].
To begin with, a positive correlation between the level of Aβ in interstitial fluid (ISF) and synaptic activity was reported in
Furthermore, various methods, such as antibodies that specifically block Aβ, knockout (KO) mice model of Aβ and its related substrates, and Aβ injection, have been implemented to actively identify the potential role of Aβ peptides involved in synaptic functions, instead of simply observing the correlation between Aβ and synaptic activity. Starting with anti-Aβ antibodies, 4G8, DAKO, and HJ5.1 were separately injected to the hippocampi of rats and mice, and a significant performance decline in behavioral tests was observed compared to controls [50, 51]. LTP, a type of neuronal activity associated with memory [52], was shown to be impaired when the endogenous Aβ was suppressed with antirodent JRF/rAb2 antibody [53]. In addition to anti-Aβ antibodies that block endogenous Aβ, a peptide called DFFVG, which binds to the receptor site of Aβ and thus blocks the activity of Aβ [54], was shown to impair learning ability as well when intracerebroventricularly injected [51]. Together, the possible involvement of Aβ in short- and long-term memory formation and learning has been suggested.
Furthermore, KO or knockdown of Aβ-related substrates in mice models was used to observe the effect of suppressing APP expression and thereby inhibiting the production of Aβ. Both KO and knockdown of APP resulted in an increased level of post-synaptic proteins, especially AMPA receptor subunit GluA1, signifying the involvement of APP in normal synaptic composition [55]. Additionally, a significant reduction in LTP was observed with knockdown of APP expression using Pen1-APP-siRNA, which can induce ~60% decline in APP expression in hippocampal cultures [53, 56], consistent with the aforementioned results shown with anti-Aβ antibodies. Similarly, conditional KO of presenilin (PS) 1 and 2, responsible for cleaving APP and thus Aβ production, also resulted in impaired synaptic plasticity and deficits in hippocampal memory
Despite the mounting evidence that suppression of Aβ production using anti-Aβ antibodies and KO/knockdown mice models resulted in impairment of LTP and memory [57-60], some researchers have expressed their concern regarding these methods to determine the physiological role of Aβ because there is a diversity of substrates and pathways that APP and the secretases can initiate and regulate [61-63]. Consequently, several research groups attempted to artificially inject synthetic Aβ into the hippocampi of mice as an alternative. Morley et al. [51] demonstrated that nanomolar concentrations of Aβ42 successfully enhanced the performance of mice in behavioral tests, signifying the enhancement of memory by Aβ. However, given that the normal physiological level of Aβ peptides has been estimated to be in picomolar concentrations [64-67], the nanomolar concentrations of Aβ administered in the study largely deviates from the endogenous level of Aβ. To address this issue, other research groups attempted to inject picomolar concentrations of Aβ, and the injected mice performed better in behavioral tests as well, confirming the enhancing effect of Aβ on synaptic plasticity and memory formation [50, 68].
Interestingly, this positive effect in low-dose Aβ directly contrasts with the inhibitive effect of high-dose Aβ against synaptic functions observed in AD patients [69], and thus it has been proposed that Aβ may operate under the mechanism of hormesis, having contrasting effects at low- and high-dose Aβ [46, 70]. Accordingly, Puzzo et al. [46, 53] identified the maximum concentrations of Aβ for the stimulatory and inhibitory hormetic responses against LTP to be 200 pM and 20 μM, respectively. Similarly, another research group demonstrated that Aβ12-28 fragment in micromolar concentrations impaired memory [71], whereas nanomolar concentrations of the same Aβ isoform resulted in a significant improvement in memory formation and learning [51]. These results suggest that the role of Aβ in regard to synaptic functions may be hormetic: inhibitory at high concentrations and stimulatory at low concentrations.
Yet, an exact isoform(s) or aggregation status of Aβ responsible for this stimulatory effect on synaptic plasticity still needs to be evaluated. In fact, some data demonstrated that it may be oligomeric Aβ42 that mediates synaptic plasticity in the hippocampus. When the production of endogenous Aβ was suppressed using anti-Aβ antibodies, 200 pM oligomeric Aβ42 rescued the impaired LTP and improved behavioral deficits, whereas a medium enriched with Aβ monomers did not have an effect [53, 72]. Also, the inhibitory effect of Aβ was only present in oligomers, while cerebral microinjection of a cell medium enriched with Aβ monomers maintained a normal LTP [10].
However, this Aβ perfusion/injection method is not without concerns. The precise concentrations of monomers and different oligomers in Aβ42 preparations perfused with each hippocampal slice cannot be accurately identified due to the propensity of the monomers to undergo conformational changes into other forms of Aβ such as dimers and oligomers [73]. In fact, when 200 pM Aβ42 oligomeric solutions were analyzed using transmission electron microscopy, the percentage of identifiable monomeric form was actually higher than that of oligomers (78.21% vs 21.78%) [72]. Also, Aβ concentration of 200 pM identified as inducing maximum enhancement in LTP [68] failed to take into an account of endogenous Aβ that the mice already had. To address these problems, perhaps novel methods or a combination of existing and new experiments could further support or reject the established hypothesis. For instance, one study devised the use of thiorphan, a competitive inhibitor to neprilysin, a presynaptic metalloprotease known to degrade endogenous Aβ, in order to increase the production of endogenous Aβ [74]. The result showed that the level of endogenous Aβ elevated in the synaptic cleft and led to overall enhancement of presynaptic strength [75]. In this fashion, more data with new experimental methods are needed to identify specific isoform(s) and aggregation status of Aβ responsible for synaptic functions.
Aside from the positive correlation reported between low concentrations of Aβ and LTP, Aβ has also been shown to target nicotinic acetylcholine receptors (nAChRs) [76, 77], which are known to promote synaptic plasticity, neuroprotection, learning and memory and regulate transmitter release in several brain regions including hippocampus [45, 78, 79]. Interestingly, mounting evidence points to the possibility that this interaction too operates under hormesis. Several studies exhibited that picomolar concentrations of Aβ42 can act as an agonist of α7-nAChRs [80, 81], resulting in increased acetylcholine production in hippocampus [51], activated PI3K [33] and elevated expression of MAP kinase, which eventually led to an increased level of LTP [82]. Additionally, another
Coupled with its hormetic interaction with nAChRs and its potential influence on presynaptic Ca2+, Aβ may conceivably interact with N-methyl-D-aspartate receptors (NMDAR) [87, 88]. Since α7-nAChR-dependent NMDAR endocytosis induces endocytosis of AMPA receptors and degradation of PSD-95, a postsynaptic scaffolding protein for synaptic development and plasticity [89], the binding of pathological Aβ at high concentrations to α7-nAChR resulted in Aβ-induced long-term depression (LTD) and synaptic dysfunction [90, 91]. Given that NMDARs can promote LTP or LTD depending on the intracellular Ca2+ concentration [92], it is possible that Aβ at picomolar concentrations may also have a physiologically normal interaction with NMDARs, though related investigations are currently lacking to reach a conclusion.
In addition to the hormetic effect of Aβ, an intriguing hypothesis on the function of Aβ on synaptic transmission has been proposed by Kamenetz et al. [93] that Aβ peptides may operate under a negative feedback loop. According to this model, the acute neuronal activity leads to an elevated production of Aβ from endogenous APP. After this increase, Aβ depresses synaptic transmission and keeps neuronal activity within a normal physiological range. Then, the level of Aβ restores back to normal. In other words, Aβ is normally maintained at an appropriate level by a negative feedback loop of Aβ, oscillating between synaptic potentiation and depression [93]. However, when this negative feedback loop of Aβ is disrupted with unknown reasons, the over-suppression of excitatory synaptic activity occurs by the accumulated Aβ and ultimately leads to the development of AD [93, 94].
Besides the physiological function of Aβ involved in synaptic plasticity and neurotransmitter receptors, a neuroprotective role of Aβ has been observed. In one study, monomeric Aβ42 activated type-1 insulin-like growth factor receptors as a positive allosteric modulator [95], which in turn induced the activation of phosphatidylinositol-3-kinase/AKT pathway [96], known to be a survival pathway for neurons [97]. Consistent with this result, Aβ monomers, especially 16~20 amino acid sequence KLVFF in the peptide, exhibited a protective activity against excitotoxic death of neurons [95, 96]. Furthermore, both synthetic and cell-derived Aβ42 monomers in nanomolar concentrations enhanced the phosphorylation of cyclic adenosine monophosphate response element binding protein (CREB), which possesses a regulative role on the expression of genes for neuronal functions such as memory and learning [98], as well as brain-derived neurotrophic factor (BDNF), which plays an important physiological role in neuronal functions and survival in normal brain [99, 100]. Taken together, these results suggest an unidentified, protective role of Aβ for cognitive and neuronal abilities by binding to insulin-like growth factor receptors and enhancing the gene expression of BDNF and CREB phosphorylation.
PROMOTION OF RECOVERY FROM BRAIN INJURY
- Go to
- Abstract
- Graphical Abstract
- INTRODUCTION
- PROTECTION AGAINST MICROBIAL INFECTIONS
- REGULATION OF SYNAPTIC FUNCTIONS
- PROMOTION OF RECOVERY FROM BRAIN INJURY
- A SEALANT FOR THE BLOOD-BRAIN BARRIER AND MODULATION OF ANGIOGENESIS
- INHIBITION OF PROLIFERATING CANCER CELLS
- Aβ-DIRECTED THERAPEUTICS IN CLINICAL TRIALS AND IMPLICATIONS
- ACKNOWLEDGEMENTS
- Figure
- Table
- Reference
Another occasion on which the level of Aβ peptides has been reported to elevate beside AD is in the case of traumatic brain injury (TBI). Accordingly, an epidemiological link between TBI and AD originating from Aβ pathology has been suggested in the past studies [101-105], though some studies have provided data against such a correlation (reviewed by Johnson et al. [106] and Tsitsopoulos et al. [107]). In fact, recent studies have reported distinct distributions of fibrillar Aβ in brains of AD and TBI patients [108] and the absence of progressive development of Aβ into plaques in long-term survivors of TBI [109]. Notably, the level of Aβ remained elevated in axons of the long-term TBI survivors without a discernable plaque formation [109]. Although their limited sample size calls for further verification to follow, the mechanisms of Aβ pathology might be different in the two diseases, possibly severing the link between AD and TBI.
Despite the unmet consensus on their association, elevated levels of Aβ in case of brain injury have consistently been published in both animal and human studies. In studies in which implemented non-transgenic mice/rodents with experimental TBI, an acute increase of rodent Aβ in damaged axons one day after injury was observed [110, 111], and even a long, persistent accumulation of Aβ was seen in some cases [112]. Interestingly, not just the levels of Aβ, but those of APP and PS1 were also restored to the normal within a week after the injury [110, 111], hinting the existence of Aβ clearance system after the initial increase. Furthermore, these results were replicated using transgenic AD model mice, engineered to express human Aβ. They exhibited elevated levels of Aβ in their axons [113-115], and the levels returned to sham levels by seven days post injury [116]. Similarly, both acute and long-term accumulations of Aβ in axons [109, 117, 118] and in cerebrospinal fluid [119-121] were confirmed in clinical trials of TBI patients, and one of the studies reported a restoration of the Aβ level to the control levels after three weeks [120], exhibiting a consistent trend with those from the animal studies. Not just a restoration of Aβ back to normal, but also fluctuation in ISF Aβ concentrations depending on neurological status of patients with acute brain injury has been observed, suggesting a potential link between neuronal activity and Aβ concentration [122]. Clearly, a positive correlation between Aβ level and brain injury exists with an undefined Aβ-clearance mechanism, and these evidence calls for a need to investigate whether Aβ is an agent for promoting recovery or a part of pathological cascade that follows after TBI [42].
In fact, some researchers have already extended their parameter to suggest a protective role of Aβ against brain injury using the BACE1 KO mouse models. However, the use of BACE1 KO mouse models rather yielded equivocal results in regard to the possible involvement of Aβ in brain injury. Loane et al. [110] reported a reduced level of cell loss and improved behavioral deficits when BACE1 was genetically inhibited in impacted mice, suggesting a deleterious role of Aβ on brain injury. Later, a contradictory result was published by Mannix and colleagues [123] that the KO of BACE1 in mice after controlled cortical impact (CCI) attenuated motor performance compared to controls. Accordingly, it has been pointed out by Mannix et al. [123] that methodological differences could be the reasons for the contrasting results. For example, Loane and associates [110] used aged (11~12 months) mice, whereas Mannix et al. [123] tested young (2~3 months) mice. Given that previous studies have suggested that the activity of BACE1 generally elevates and TBI worsens with aging [124, 125], an age-dependent effect of TBI on Aβ is also a possibility and thus call for further studies. Additionally, a detrimental effect of BACE1 KO was observed within a week in the study by Mannix et al. [123], whereas a beneficial effect of inhibition of BACE1 was only apparent 2~3 weeks after CCI, rendering a direct comparison rather difficult. Nonetheless, Mannix and colleagues published a follow-up study showing a rescuing effect of intracerebroventricularly injected Aβ40 against impaired motor memory in TBI-impacted BACE1-/- mice, highlighting a protective effect of Aβ [126]. Moreover, another evidence in support of the beneficial effect of Aβ in brain injury has been provided by Pajoohesh-Ganji et al., as reduced level of endogenous Aβ achieved using γ-secretase inhibitor DAPT or BACE1 KO mice exhibited attenuated functional recovery after spinal cord injury (SCI) [111].
However, the results from BACE1-/- rat models need to be interpreted with caution because other substrates of BACE1, instead of Aβ, could be responsible for the observed effect. In fact, several studies have shown that the inhibition of BACE1 either by KO mouse model or its inhibitors led to an elevated level of axonal regeneration after injury, and vice versa for the elevated BACE expression [127-129]. Also, inhibiting BACE1 in both axons and Schwann cells of injured rats resulted in a reduced level of remyelination [130, 131], a process significant for restoring the function of demyelinated axons from brain damage [132]. Nonetheless, these results have been concluded to be independent of Aβ; neuregulin-1, one of the substrates of BACE1, instead is likely to be responsible for the regulation of remyelination [133, 134]. Likewise, other substrates of BACE1, such as Jagged-1 and Jagged-2 [135], might affect the results from the BACE-/- model. Hence, further investigations are in need to identify the specific substrate(s) that is responsible for the observed effect of BACE1 KO on brain injury.
Aside from TBI, focal cerebral ischemia, which results in occlusion of blood vessels in brain, was also found to be correlated with Aβ accumulations [136, 137], and these findings open up a possibility that Aβ may have a functional role against other types of brain injury as well. One study demonstrated that a transgenic mouse model that overexpresses human APP (
Also, the effect of Aβ against experimental autoimmune encephalomyelitis (EAE), an inflammatory disease in brain, was tested by Grant et al. [139]. Their results from the intraperitoneal administration of Aβ40 and Aβ42 enriched in monomers and oligomers demonstrated that the treatment before the onset of the disease successfully decreased the number of incidence and severity of the disease, and the administration of Aβ after the onset diminished motor paralysis compared to the control group. In addition, Aβ was also shown effective against EAE accelerated by proinflammatory T helper cells (TH1 and TH17) as it significantly decreased the severity of the disease and prevented the production of TH1 and TH17 related proinflammatory cytokines such as IL-6, IFN-γ, and IL-17 [139]. Notably, this protective effect of Aβ was achieved without Aβ localizing in the CNS.
Although increasing evidence on the positive effect of Aβ in the cases of brain injury is available, conflicting results concerning the relationship between Aβ accumulation and TBI still call for a thorough evaluation of their limitations and further investigation. To begin with, many studies in the past have used transgenic mouse models with a genetic predisposition to human familial AD via overexpression of human APP and/or PS1/2 to prove a causal link between TBI and AD. However, the use of these models failed to consider the possibility that the overexpression of the transgenes followed by the accumulation of Aβ and pathological deficits from AD might have influenced the acquired outcomes. Specifically, it is hard to determine which came first: did a genetic predisposition to AD exacerbate TBI or did TBI lead to AD? Accordingly, a recent study led by Maigler and colleagues employed a transgenic mouse model of non-mutated, human APP with KO of the Aβ-degrading enzyme neprilysin (APPtg.NEP-/-) as an alternative to the conventional AD models and examined the effect of reduced Aβ clearance in TBI [140]. Consistent with the aforementioned studies conducted with the transgenic AD mice, the APPtg.NEP-/- mice demonstrated an elevated level of Aβ 1 day after TBI. Notably, the neprilysin-deficient mice performed better in behavioral tests compared to wild-type mice, and no significant difference was observed between impacted and non-impacted neprilysin-deficient mice [140]. Hence, the authors argued a protective role of Aβ against brain injury. Although it remains to be investigated whether it was Aβ or other substrates of neprilysin that were responsible for this positive effect, this normalized human APP mouse model could be used as an alternative for future studies on the effect of Aβ against brain injury.
In addition to the limitation of the use of AD mouse models, methodological inconsistencies among studies have been a primary drawback of animal studies examining the level of cerebral Aβ after TBI (reviewed by Bird et al. [141]). For instance, the animal models used have ranged from transgenic ones expressing human familial AD mutations such as APPswe and PS-1swe to non-transgenic ones such as CD1. Undoubtedly, more clinically relevant data would be obtained with transgenic mice with a normal level of human Aβ due to its difference with rodent Aβ [142-144]. Also, the surgical procedures used to incur TBI differed among studies, leading to various severities of TBI and diverse regions of brain being affected. For example, a contradictory result was observed when rats were subjected to blast overpressure exposure instead of controlled cortical impact, whereby the level of endogenous rodent Aβ decreased [145]. Additionally, variability in the ages of rats, durations of experiments, and types of Aβ assay used such as ELISA and IHC have made it difficult to directly compare the results of different studies.
However, it is not just animal studies that encompasses limitations; clinical results from TBI patients are also not so free of similar limitations. Limited sample sizes and heterogeneity of TBI in each patient have always hindered researchers from reaching a concrete conclusion. For example, while one study has reported no correlations between the ISF Aβ level and the level of consciousness in TBI patients [117], the data from another study has shown that the ISF Aβ correlated with the neurological status of their patients [122]. Therefore, more concrete evidence is needed to confirm the physiological role of Aβ in brain injury.
A SEALANT FOR THE BLOOD-BRAIN BARRIER AND MODULATION OF ANGIOGENESIS
- Go to
- Abstract
- Graphical Abstract
- INTRODUCTION
- PROTECTION AGAINST MICROBIAL INFECTIONS
- REGULATION OF SYNAPTIC FUNCTIONS
- PROMOTION OF RECOVERY FROM BRAIN INJURY
- A SEALANT FOR THE BLOOD-BRAIN BARRIER AND MODULATION OF ANGIOGENESIS
- INHIBITION OF PROLIFERATING CANCER CELLS
- Aβ-DIRECTED THERAPEUTICS IN CLINICAL TRIALS AND IMPLICATIONS
- ACKNOWLEDGEMENTS
- Figure
- Table
- Reference
Among different hypotheses for AD pathogenesis, ‘the vascular hypothesis’ states that acute changes in cerebral vasculature system such as disrupted blood-brain barrier (BBB) and neoangiogenesis could lead to AD, and amyloidosis has been suspected to trigger such an event [146-148]. However, there is emerging evidence that non-pathological Aβ can in fact regulate angiogenesis, an essential physiological process for developing new blood vessels in cases of developing organs, healing wound, and so on [149], and potentially protect leakages in blood brain barrier (BBB) in a hormetic manner [42, 150, 151]. Several studies on physiochemical properties of Aβ and its localization to injured cerebrovascular regions suggest that Aβ peptides may serve as a protective ‘scab’, in other words a sealant, that maintains the integrity of the BBB and seals its leakages (reviewed by Atwood et al. [150, 152]. These leakages are typical of cerebrovascular alterations such as decreased levels of endothelial cells and microvascular density found in both aged people and AD patients [153-156]. Notably, it has also been pointed out that cerebral microhemorrhage observed when deposited Aβ was removed by immunotherapy in a cerebral amyloid angiopathy mouse model is evidence that Aβ has a role in maintaining vascular integrity [157]. Another
Still, more research on the physiological role of Aβ in regard to angiogenesis and sealing BBB is in need because the general aim of previous studies has been finding the potential relation of Aβ-induced neoangiogenesis to AD pathology, rather than elucidating the sole effect of Aβ peptide. For example, Biron et al. [147] reported hypervascularity in transgenic AD mice (Tg2565) in 18- to 24-month-old, and another study has shown a positive correlation between vascular density in the hippocampus and Aβ loads in postmortem AD brain tissues using immunoactivity of αvβ3, a marker for angiogenesis [160].
Interestingly, in line with the previously mentioned studies indicating the hormetic effect of Aβ on several physiological functions, a series of
In addition to the dose-dependent effect of Aβ, a conformation-dependent effect on angiogenesis has also been observed. The oligomeric form of Aβ peptides exhibited an anti-angiogenic activity, while fibrillar forms did not shown to be active in the same assay [170]. This result was further supported by the follow-up study that the amino acid sequence identified to be responsible for the anti-angiogenic effect is HHQKLVFF, of which includes the LVFF sequence known to be exposed in the oligomeric form of Aβ [171]. Conversely, a pro-angiogenic effect was seen in Aβ35-42, a motif that may protrude outwards in monomeric or dimeric states with its property of solvent-inaccessibility [171, 172]. Another study showed that incubation of Aβ42 monomers in 225 nM with human umbilical vein endothelial cells resulted in an elevated number of endothelial tip cell formation without any apparent apoptotic death [167]. Together, these results signify a potentially non-pathological effect of monomeric Aβ on angiogenesis.
INHIBITION OF PROLIFERATING CANCER CELLS
- Go to
- Abstract
- Graphical Abstract
- INTRODUCTION
- PROTECTION AGAINST MICROBIAL INFECTIONS
- REGULATION OF SYNAPTIC FUNCTIONS
- PROMOTION OF RECOVERY FROM BRAIN INJURY
- A SEALANT FOR THE BLOOD-BRAIN BARRIER AND MODULATION OF ANGIOGENESIS
- INHIBITION OF PROLIFERATING CANCER CELLS
- Aβ-DIRECTED THERAPEUTICS IN CLINICAL TRIALS AND IMPLICATIONS
- ACKNOWLEDGEMENTS
- Figure
- Table
- Reference
Despite the notorious notion that AD encompasses, meta-analyses have demonstrated that AD patients have lower risks of developing various types of cancers such as bladder, breast, lung, colorectal, head and neck cancers and hematologic malignancies compared to cognitively normal elderly individuals [173-176]. This notable negative correlation drew researchers’ attention to Aβ, and it has been hypothesized that Aβ may inhibit the growth of tumor cells [164, 177, 178]. In support of this hypothesis, a direct injection of freshly dissolved Aβ into human lung adenocarcinoma xenografts and human glioblastoma suppressed the tumor growth in nude mice [164]. In addition to nude mice, transgenic mice that overexpress Aβ exhibited 50% of reduction in tumor volume compared to wild-type when glioma tumors were intracranially injected [177]. Consistent result was observed with cell-derived Aβ from mammalian cells as well, preventing the proliferation of human glioblastoma cells, breast cancer cells and mouse skin cancer cells in a concentration-dependent manner [178].
Here, a potential mechanism proposed for the antitumor effect of Aβ is by promoting apoptosis. A couple of studies indicated that APP partially mediated the expression of p53, a gene responsible for controlling apoptosis [179] and that Aβ42 bound to the promotor of p53, eventually increasing the rate of transcription of p53 [180]. Also, high nanomolar concentrations of Aβ42 increased susceptibility to oxidative stress and resulted in a lower expression of X-linked inhibitor of apoptosis (XIAP), leading to downregulation of XIAP, which directly inhibits key proteases of the apoptosis cascade, such as caspase 3, 7 and 9 [181, 182]. Not limited to p53 and XIAP, but Bcl-2, a vital anti-apoptotic protein, was also shown to be negatively influenced by Aβ42, while the level of Bax, which promotes cell apoptosis, increased when 100 nM of Aβ42 was added to primary neuron cultures [183]. This result is noteworthy because Bcl-2 overexpression is commonly observed in many types of cancer [184], further highlighting the hypothesis that Aβ may contribute to reducing cancer risks by promoting apoptosis.
Despite the growing evidence, the antitumor role of Aβ is not conclusive due to limited data and methodology. In the aforementioned studies on the inhibitive effect of Aβ on the growth of tumor cells, either freshly solubilized Aβ or naturally secreted soluble Aβ were incubated with cancer cells, without further identifying the exact species, monomeric or further aggregated form, responsible for the observed effect [164, 177, 178]. Therefore, more research on a major contributor of Aβ species to the potential antitumor role of Aβ is required.
Aβ-DIRECTED THERAPEUTICS IN CLINICAL TRIALS AND IMPLICATIONS
- Go to
- Abstract
- Graphical Abstract
- INTRODUCTION
- PROTECTION AGAINST MICROBIAL INFECTIONS
- REGULATION OF SYNAPTIC FUNCTIONS
- PROMOTION OF RECOVERY FROM BRAIN INJURY
- A SEALANT FOR THE BLOOD-BRAIN BARRIER AND MODULATION OF ANGIOGENESIS
- INHIBITION OF PROLIFERATING CANCER CELLS
- Aβ-DIRECTED THERAPEUTICS IN CLINICAL TRIALS AND IMPLICATIONS
- ACKNOWLEDGEMENTS
- Figure
- Table
- Reference
Given the potential clinical significance and market effects of AD therapeutics, there have been many attempts to target Aβ using various approaches focusing on different stages of the amyloid cascade (Table 1) [185-228]. For example, inhibitors of Aβ-related secretases aim to decrease the production of Aβ itself as a preventative measure, while anti-Aβ immunotherapies induce partial or complete clearance of Aβ at the end of the amyloid cascade. However, a lack of efficiency and unexpected adverse side effects have hampered AD patients from receiving effective treatment and led to a reliance on drugs such as donepezil (AChE inhibitor) and memantine (NMDA receptor antagonist) that are designed to temporarily alleviate symptoms [229]. Recently, the US Food and Drug Administration (FDA) granted approval for aducanumab as the first disease-modifying therapy for AD; however, many professionals expressed concerns and criticism of this decision due to insufficient data showing the effectiveness of the drug on rescuing the loss of cognitive abilities. Therefore, an important question must be answered: why are Aβ-targeted drugs failing?
To begin with, the earliest components of the amyloid cascade targeted by AD therapeutics are BACE1- and γ- secretases by their respective inhibitors. Co-cleavage of APP by BACE1- and γ- secretases is known to release Aβ into the extracellular space; inhibition of these secretases has therefore been proposed as a measure to block the Aβ production, regardless of the conformation of Aβ. However, the clinical outcomes of both BACE1- and γ- secretases have been disappointing because many were discontinued due to a lack of efficiency and worsen cognition among the tested patients compared to placebo group (Table 1). For example, a series of oral BACE1 inhibitors, including Verubecestat [185], Lanabecestat [189], and Atabecestat [230], which underwent clinical trials on mild AD patients were shown to exacerbate cognitive functions of the participants, despite the apparent reduction in CSF Aβ levels. Also, the clinical trials of γ- secretase inhibitors, Semagacestat [194] and Avagacestat [4, 196] were halted as their administration resulted in aggravated functional and cognitive abilities, and higher rates of skin cancer and infections [231]. These adverse events observed in the clinical trials of BACE1- and γ- secretase inhibitors were consistent with
In response to the apparent failure in the upstream regulation of the amyloid cascade via the secretase inhibitors, more recent attempts have been made with anti-Aβ immunotherapies. These immunization methods are intended to reduce the level of Aβ deposits in the brain through either an active approach using Aβ antigen that generate an antibody response against Aβ or a passive approach using humanized Aβ antibodies [232]. An initial attempt was made by Elan Pharmaceuticals with the AN-1792 Aβ antigen, which is a synthetic, aggregated Aβ42 combined with adjuvant, and the result showed a significant reduction in Aβ loads in the participants. Nonetheless, no improvement in their cognitive decline was observed [198, 233]. Notably, ~6% of the immunized patients exhibited meningoencephalitis, which is an inflammatory disease of the brain caused by infection [198, 234]. This unexpected adverse effect suggests that there may be a need to keep normal forms of Aβ rather than complete clearance, because it has been suggested that monomeric and oligomeric Aβ may exert an antimicrobial effect and have a protective role against brain inflammatory diseases by suppression of the production of proinflammatory cytokines [139]. Consequently, lack of clinical efficiency and adverse side effects such as cognitive worsening in active immunotherapies such as AD02 [200] and CAD106 [202] that followed after AN-1792 turned many researchers to move onto the passive Aβ immunotherapy.
Similarly, the passive immunotherapy aims to clear out Aβ using monoclonal or polyclonal anti-Aβ antibodies targeting neurotoxic Aβ. Among the antibodies tested, aducanumab by Biogen has received the greatest attention because of successful phase I results indicating the dose- and time-dependent clearance of Aβ plaques on positron emission tomography scans and slowed progressive cognitive decline of AD patients [204]. Aducanumab was the first such drug to be approved by the FDA as an AD therapeutic. However, many professionals, even some advisory members of the FDA, have criticized the approval of aducanumab due to inadequate evidence for clinical effects in the phase III trials (EMERGE and ENGAGE) and due to the subjectivity of the FDA as they worked with the sponsor to reevaluate the submitted data [235-237].
Adding on to the ambiguous findings for aducanumab and the apparent lack of efficiency of other Aβ immunotherapies, a new adverse effect, amyloid-related imaging abnormalities (ARIA), has been reported in clinical trials of a significant number of the immunized Aβ antibodies such aducanumab [203], bapineuzumab [206], gantenerumab [238], and solanezumab [15] (reviewed by DiFrancesco et al. [239]). Based on radiographic features of the subjects, ARIA is divided into two subgroups: ARIA-E if vasogenic edema is observed and ARIA-H by microhemorrhages with hemosiderin deposits in brain [240]. Interestingly, the subjects were either symptomatic with several neurological side effects or asymptomatic in some cases [203, 205]. One hypothesis suggested for pathophysiology of ARIA is the solubilization of parenchymal Aβ plaques by the immunotherapies leading to the accumulation of cerebrovascular Aβ in vessel walls. Then, this accumulation is hypothesized to be stimulating the clearance of vascular Aβ by relative lymphocytes and proteases and weakening of BBB, and thus edema and hemorrhages are spotted [240-242]. Although very limited evidence for risk factors and mechanisms of ARIA is available so far, any signs of ARIA in the brain of the patients must be carefully screened in future trials.
Continuous failures of the Aβ-targeting therapeutics and adverse effects highlight the need for further elucidation of the pathogenesis of AD and for the adjustment on AD therapeutics. One hypothesis to explain the lack of efficiency of the Aβ immunotherapies is that the AD patients in clinical trials were too advanced in their stage of AD and thus no longer exhibited the clinical effect observed in the preclinical stages [243]. It is possible that the antibodies may be effective only as a preventative measure, and do not engage with Aβ plaques once profound abnormalities in neuropathology have developed. Additionally, an increased level of soluble oligomeric Aβ after the clearance of Aβ plaques by aducanumab has been proposed as another possibility for the failure, as mounting evidence points to deleterious effects of diffusible Aβ oligomers [244]. The possibility that Aβ might not be a desirable target for clinical interventions in AD should also be considered; indeed, a recent meta-analysis of randomized trials of Aβ-directed drugs exhibited that Aβ clearance therapies did not cause a significant improvement in cognition [245]. Based on these considerations, many new approaches for AD therapeutics have been suggested, including modifications of targeted epitopes of Aβ, adjustments on administered doses, and direct-delivery methods across the BBB [246].
Based on the physiological roles of Aβ discussed earlier (Fig. 1), novel therapeutics for AD could be designed to selectively remove or disaggregate neurotoxic forms of Aβ while retaining its monomeric form in the brain. We hypothesize that the side effects reported in the clinical trials of Aβ-targeting drug candidates might have been resulted from the complete clearance of Aβ from the brain, which led to the unexpected loss of monomeric Aβ and their functions. For instance, the weakened BBB evident through ARIA might be related to normal Aβ peptides no longer being able to work as a protective scab for BBB. Also, the patients with reduced Aβ could have been more susceptible to microbial infections with reduced number of Aβ monomers with antimicrobial activity, which could explain the increased infection rates observed in the clinical trials. More importantly, the absence of monomeric Aβ that has been shown to regulate synaptic activity could be the reason why many subjects with reduced Aβ levels experienced worsened cognition in the trials. Although more direct evidence is required to verify this hypothesis, the restoration of monomeric Aβ in the brain has a potential to elicit cognitive improvement and satisfy the unmet clinical need for AD therapeutics.
ACKNOWLEDGEMENTS
- Go to
- Abstract
- Graphical Abstract
- INTRODUCTION
- PROTECTION AGAINST MICROBIAL INFECTIONS
- REGULATION OF SYNAPTIC FUNCTIONS
- PROMOTION OF RECOVERY FROM BRAIN INJURY
- A SEALANT FOR THE BLOOD-BRAIN BARRIER AND MODULATION OF ANGIOGENESIS
- INHIBITION OF PROLIFERATING CANCER CELLS
- Aβ-DIRECTED THERAPEUTICS IN CLINICAL TRIALS AND IMPLICATIONS
- ACKNOWLEDGEMENTS
- Figure
- Table
- Reference
This research was supported by the Korea Health Technology R&D Project (Grant Number: HU21C0161) through the Korea Health Industry Development Institute (KHIDI) and Korea Dementia Research Center (KDRC), and Mid-Career Researcher Program (Grant Number: NRF-2021R1A2C2093916), and Basic Science Research Program (Grant Number: NRF-2018R1A6A1A03023718) through the National Research Foundation of Korea (NRF), funded by the Ministry of Health & Welfare and Ministry of Science and ICT, Republic of Korea. This research was also supported by Yonsei Frontier Lab, Amyloid Solution, and POSCO Science Fellowship of POSCO TJ Park Foundation.
Figures
- Go to
- Abstract
- Graphical Abstract
- INTRODUCTION
- PROTECTION AGAINST MICROBIAL INFECTIONS
- REGULATION OF SYNAPTIC FUNCTIONS
- PROMOTION OF RECOVERY FROM BRAIN INJURY
- A SEALANT FOR THE BLOOD-BRAIN BARRIER AND MODULATION OF ANGIOGENESIS
- INHIBITION OF PROLIFERATING CANCER CELLS
- Aβ-DIRECTED THERAPEUTICS IN CLINICAL TRIALS AND IMPLICATIONS
- ACKNOWLEDGEMENTS
- Figure
- Table
- Reference

{kind=link}
Tables
- Go to
- Abstract
- Graphical Abstract
- INTRODUCTION
- PROTECTION AGAINST MICROBIAL INFECTIONS
- REGULATION OF SYNAPTIC FUNCTIONS
- PROMOTION OF RECOVERY FROM BRAIN INJURY
- A SEALANT FOR THE BLOOD-BRAIN BARRIER AND MODULATION OF ANGIOGENESIS
- INHIBITION OF PROLIFERATING CANCER CELLS
- Aβ-DIRECTED THERAPEUTICS IN CLINICAL TRIALS AND IMPLICATIONS
- ACKNOWLEDGEMENTS
- Figure
- Table
- Reference
A list of therapeutic approaches to target Aβ and their adverse events in clinical trials
| Mechanism of action | Drug | Trial phase | Adverse events in clinical trials | Main reason for failure | References |
|---|---|---|---|---|---|
| BACE1 inhibitor | Verubecestat | III | Worsened cognition, increased mortality, and reduced brain volume in the whole-brain and hippocampal region | Lack of efficacy | Egan et al. (2018) [185], Sur et al. (2020) [186] |
| Umibecestat (CNP520) | II / III | Worsened cognition and brain atrophy | Toxicity | NCT02565511, NCT03131453 | |
| Atabecestat | II / III | Worsened cognition and liver toxicity | Toxicity | Novak et al. (2020) [187], Sperling et al. (2021) [188] | |
| Lanabecestat (AZD3293, LY3314814) | III | Worsened cognition | Lack of efficacy | Wessels et al. (2020) [189] | |
| BI1181181 | I | No SAEs reported | Further study required | NCT02044406, Nicolas et al. (2015) [190] | |
| LY2886721 | II | Liver toxicity | Toxicity | Lahiri et al. (2014) [191] | |
| AZD3839 | I | Heart rhythm disturbance | Toxicity | Yan (2016) [192], NCT01348737 | |
| RG7129 | I | Liver toxicity | Toxicity | NCT01664143, NCT01592331 | |
| Elenbecestat (E2609) | III | No SAEs reported | Lack of efficacy | NCT02956486, NCT03036280 | |
| LY3202626 | II | No SAEs reported | Lack of efficacy | Lo et al. (2021) [193] | |
| γ-secretase inhibitor | Semagacestat | III | Worsened cognition and increased skin cancers and infections | Toxicity and lack of efficacy | Doody et al. (2013) [194] |
| Begacestat (GSI-953) | I | No SAEs reported | Further study required | NCT00547560 | |
| Avagacestat | II | Worsened cognition and increased nonmelanoma skin cancer | Toxicity and lack of efficacy | Coric et al. (2012) [195], Coric et al. (2015) [196] | |
| MPC-7869 (Flurizan, Tarenflurbil) | III | No SAEs reported | Lack of efficacy | NCT00322036 Green et al. (2009) [197] | |
| Aβ antigen | AN-1792 (AIP001) | II | Brain inflammation (meningoencephalitis) | Toxicity and lack of efficacy | Gilman et al. (2005) [198], Boche et al. (2010) [199] |
| Affitope AD02 | II | No SAEs reported | Lack of efficacy | Schneeberger et al. (2015) [200] | |
| Vanutide cridificar (ACC-001) | II | No SAEs reported | Lack of efficacy | Pasquier et al. (2016) [201] | |
| CAD 106 (Amilomotide) | II | Worsened cognition, decreased cortical gray-matter volume, and ARIAs (ARIA-E & ARIA-H) | Lack of efficacy | Vandenberghe et al. (2017) [202] | |
| Monoclonal antibody | Aducanumab (BIIB037, Aduhelm) | III | ARIA-E | Lack of efficacy | Ferrero et al. (2016) [203], Sevigny et al. (2016) [204], NCT02477800, NCT02484547 |
| Bapineuzumab (AAB-001) | III | Increased risk of serious treatment-emergent adverse events and ARIA-E | Toxicity and Lack of efficacy | Salloway et al. (2014) [205], Vandenberghe et al. (2016) [206], Abushouk et al. (2017) [207] | |
| AAB-003 | I | ARIA-E | Further study required | Delnomdedieu et al. (2016) [208], NCT01193608 | |
| Gantenerumab | III | ARIAs | Lack of efficacy | Ostrowitzki et al. (2017) [209] | |
| Solanezumab (LY2062430) | III | ARIAs | Lack of efficacy | Doody et al. (2014) [210], Honig et al. (2018) [15], Siemers et al. (2016) [211] | |
| Crenezumab | III | No SAEs reported | Lack of efficacy | Yang et al. (2019) [212], NCT02670083 | |
| Ponezumab | II | No SAEs reported | Lack of efficacy | Landen et al. (2017) [213], NCT00722046 | |
| Immunoglobin | III | No SAEs reported | Lack of efficacy | Relkin et al. (2017) [214] | |
| Donanemab (LY3002813) | III | Reduced brain volume and ARIA-E | Lack of efficacy | Lowe et al. (2021) [215], Ayton (2021) [216] NCT04437511 | |
| Lecanemab (BAN2401) | II | ARIA-E | Lack of efficacy | Swanson et al. (2021) [217] NCT03887455 | |
| SAR228810 | I | No SAEs reported | Further study required | Pradier et al. (2018) [218], NCT01485302 | |
| MEDI1814 | I | No SAEs reported | Further study required | NCT02036645 | |
| GSK933776 | II | No SAEs reported | Lack of efficacy | Leyhe et al. (2014) [219], NCT01342926 | |
| Aβ vaccine | ACI-24 | II | No SAEs reported | Further study required | Ritchie et al. (2003) [220] |
| UB-311 | II | No SAEs reported | Further study required | Wang et al. (2017) [221], NCT02551809 | |
| ABVac40 | II | No SAEs reported | Further study required | Lacosta et al. (2018) [222] NCT03461276 | |
| Aβ aggregation inhibitor | Alzhemed (Tramiprosate, 3-APS) | III | No SAEs reported | Lack of efficacy | NCT00314912, Aisen et al. (2006) [223], Gauthier et al. (2009) [224] |
| Scyllo-inositol (AZD-103, ELND005) | II | Higher incidence of SAEs and respiratory tract infections in high dose groups | Lack of efficacy, Further study required | Salloway et al. (2011) [225] | |
| PBT1 (Clioquinol) | II | SAEs such as visual impairment and intracranial hemorrhage | Toxicity and lack of efficacy | Sampson et al. (2014) [226] | |
| PBT2 (Hydroxyquinoline) | II | No SAEs reported | Lack of efficacy | Lannfelt et al. (2008) [227] NCT00471211 | |
| GV-971 (Sodium oligo-mannurarate) | III | Higher incidence of hyperlipidemia and nasopharyngitis | Further study required | Xiao et al. (2021) [228] NCT02293915 |
SAE, serious adverse events; ARIA-E, amyloid-related imaging abnormalities with vasogenic edema; ARIA-H, amyloid-related imaging abnormalities with microhemorrhages.
References
- Go to
- Abstract
- Graphical Abstract
- INTRODUCTION
- PROTECTION AGAINST MICROBIAL INFECTIONS
- REGULATION OF SYNAPTIC FUNCTIONS
- PROMOTION OF RECOVERY FROM BRAIN INJURY
- A SEALANT FOR THE BLOOD-BRAIN BARRIER AND MODULATION OF ANGIOGENESIS
- INHIBITION OF PROLIFERATING CANCER CELLS
- Aβ-DIRECTED THERAPEUTICS IN CLINICAL TRIALS AND IMPLICATIONS
- ACKNOWLEDGEMENTS
- Figure
- Table
- Reference
- Spires-Jones TL, Hyman BT (2014) The intersection of amyloid beta and tau at synapses in Alzheimer's disease. Neuron 82:756-771



- Masliah E, Mallory M, Alford M, DeTeresa R, Hansen LA, McKeel DW Jr, Morris JC (2001) Altered expression of synaptic proteins occurs early during progression of Alzheimer's disease. Neurology 56:127-129
- Shankar GM, Walsh DM (2009) Alzheimer's disease: synaptic dysfunction and Abeta. Mol Neurodegener 4:48
- Bishop GM, Robinson SR (2002) The amyloid hypothesis: let sleeping dogmas lie?. Neurobiol Aging 23:1101-1105
- Storey E, Cappai R (1999) The amyloid precursor protein of Alzheimer's disease and the Abeta peptide. Neuropathol Appl Neurobiol 25:81-97
- Hardy J, Selkoe DJ (2002) The amyloid hypothesis of Alzheimer's disease: progress and problems on the road to therapeutics. Science 297:353-356
- Ricciarelli R, Fedele E (2017) The amyloid cascade hypothesis in Alzheimer's disease: it's time to change our mind. Curr Neuropharmacol 15:926-935
- Cleary JP, Walsh DM, Hofmeister JJ, Shankar GM, Kuskowski MA, Selkoe DJ, Ashe KH (2005) Natural oligomers of the amyloid-beta protein specifically disrupt cognitive function. Nat Neurosci 8:79-84
- Shankar GM, Bloodgood BL, Townsend M, Walsh DM, Selkoe DJ, Sabatini BL (2007) Natural oligomers of the Alzheimer amyloid-beta protein induce reversible synapse loss by modulating an NMDA-type glutamate receptor-dependent signaling pathway. J Neurosci 27:2866-2875
- Walsh DM, Klyubin I, Fadeeva JV, Cullen WK, Anwyl R, Wolfe MS, Rowan MJ, Selkoe DJ (2002) Naturally secreted oligomers of amyloid beta protein potently inhibit hippocampal long-term potentiation in vivo. Nature 416:535-539
- Uddin MS, Kabir MT, Rahman MS, Behl T, Jeandet P, Ashraf GM, Najda A, Bin-Jumah MN, El-Seedi HR, Abdel-Daim MM (2020) Revisiting the amyloid cascade hypothesis: from anti-Aβ therapeutics to auspicious new ways for Alzheimer's Disease. Int J Mol Sci 21:5858
- Jeremic D, Jiménez-Díaz L, Navarro-López JD (2021) Past, present and future of therapeutic strategies against amyloid-β peptides in Alzheimer's disease: a systematic review. Ageing Res Rev 72:101496
- Farlow M, Arnold SE, van Dyck CH, Aisen PS, Snider BJ, Porsteinsson AP, Friedrich S, Dean RA, Gonzales C, Sethuraman G, DeMattos RB, Mohs R, Paul SM, Siemers ER (2012) Safety and biomarker effects of solanezumab in patients with Alzheimer's disease. Alzheimers Dement 8:261-271
- Egan MF, Kost J, Voss T, Mukai Y, Aisen PS, Cummings JL, Tariot PN, Vellas B, van Dyck CH, Boada M, Zhang Y, Li W, Furtek C, Mahoney E, Harper Mozley L, Mo Y, Sur C, Michelson D (2019) Randomized trial of verubecestat for prodromal Alzheimer's disease. N Engl J Med 380:1408-1420
- Honig LS, Vellas B, Woodward M, Boada M, Bullock R, Borrie M, Hager K, Andreasen N, Scarpini E, Liu-Seifert H, Case M, Dean RA, Hake A, Sundell K, Poole Hoffmann V, Carlson C, Khanna R, Mintun M, DeMattos R, Selzler KJ, Siemers E (2018) Trial of solanezumab for mild dementia due to Alzheimer's disease. N Engl J Med 378:321-330
- Moore DB, Gillentine MA, Botezatu NM, Wilson KA, Benson AE, Langeland JA (2014) Asynchronous evolutionary origins of Aβ and BACE1. Mol Biol Evol 31:696-702
- Robinson SR, Bishop GM (2002) Abeta as a bioflocculant: implications for the amyloid hypothesis of Alzheimer's disease. Neurobiol Aging 23:1051-1072
- Moir RD, Lathe R, Tanzi RE (2018) The antimicrobial protection hypothesis of Alzheimer's disease. Alzheimers Dement 14:1602-1614
- Zaiou M (2007) Multifunctional antimicrobial peptides: therapeutic targets in several human diseases. J Mol Med (Berl) 85:317-329
- Pastore A, Raimondi F, Rajendran L, Temussi PA (2020) Why does the Aβ peptide of Alzheimer share structural similarity with antimicrobial peptides?. Commun Biol 3:135
- Lee EY, Srinivasan Y, de Anda J, Nicastro LK, Tükel Ç, Wong GCL (2020) Functional reciprocity of amyloids and antimicrobial peptides: rethinking the role of supramolecular assembly in host defense, immune activation, and inflammation. Front Immunol 11:1629
- Zanetti M (2004) Cathelicidins, multifunctional peptides of the innate immunity. J Leukoc Biol 75:39-48
- Soscia SJ, Kirby JE, Washicosky KJ, Tucker SM, Ingelsson M, Hyman B, Burton MA, Goldstein LE, Duong S, Tanzi RE, Moir RD (2010) The Alzheimer's disease-associated amyloid beta-protein is an antimicrobial peptide. PLoS One 5:e9505
- Kumar DK, Choi SH, Washicosky KJ, Eimer WA, Tucker S, Ghofrani J, Lefkowitz A, McColl G, Goldstein LE, Tanzi RE, Moir RD (2016) Amyloid-β peptide protects against microbial infection in mouse and worm models of Alzheimer's disease. Sci Transl Med 8:340ra72
- White MR, Kandel R, Tripathi S, Condon D, Qi L, Taubenberger J, Hartshorn KL (2014) Alzheimer's associated β-amyloid protein inhibits influenza A virus and modulates viral interactions with phagocytes. PLoS One 9:e101364
- Eimer WA, Vijaya Kumar DK, Navalpur Shanmugam NK, Rodriguez AS, Mitchell T, Washicosky KJ, György B, Breakefield XO, Tanzi RE, Moir RD (2018) Alzheimer's disease-associated β-amyloid is rapidly seeded by herpesviridae to protect against brain infection. Neuron 99:56-63.e3
- Bourgade K, Garneau H, Giroux G, Le Page AY, Bocti C, Dupuis G, Frost EH, Fülöp T Jr (2015) β-Amyloid peptides display protective activity against the human Alzheimer's disease-associated herpes simplex virus-1. Biogerontology 16:85-98
- Spitzer P, Condic M, Herrmann M, Oberstein TJ, Scharin-Mehlmann M, Gilbert DF, Friedrich O, Grömer T, Kornhuber J, Lang R, Maler JM (2016) Amyloidogenic amyloid-β-peptide variants induce microbial agglutination and exert antimicrobial activity. Sci Rep 6:32228
- Tripathi S, Tecle T, Verma A, Crouch E, White M, Hartshorn KL (2013) The human cathelicidin LL-37 inhibits influenza A viruses through a mechanism distinct from that of surfactant protein D or defensins. J Gen Virol 94(Pt 1):40-49
- Lukiw WJ, Cui JG, Yuan LY, Bhattacharjee PS, Corkern M, Clement C, Kammerman EM, Ball MJ, Zhao Y, Sullivan PM, Hill JM (2010) Acyclovir or Aβ42 peptides attenuate HSV-1-induced miRNA-146a levels in human primary brain cells. Neuroreport 21:922-927
- Güntert A, Döbeli H, Bohrmann B (2006) High sensitivity analysis of amyloid-beta peptide composition in amyloid deposits from human and PS2APP mouse brain. Neuroscience 143:461-475
- White MR, Kandel R, Hsieh IN, De Luna X, Hartshorn KL (2018) Critical role of C-terminal residues of the Alzheimer's associated β-amyloid protein in mediating antiviral activity and modulating viral and bacterial interactions with neutrophils. PLoS One 13:e0194001
- Luo Y, Sunderland T, Roth GS, Wolozin B (1996) Physiological levels of beta-amyloid peptide promote PC12 cell proliferation. Neurosci Lett 217:125-128
- Wiatrak B, Balon K (2021) Protective activity of Aβ on cell cultures (PC12 and THP-1 after differentiation) preincubated with lipopolysaccharide (LPS). Mol Neurobiol 58:1453-1464
- Iqbal UH, Zeng E, Pasinetti GM (2020) The use of antimicrobial and antiviral drugs in Alzheimer's disease. Int J Mol Sci 21:4920
- Spitzer P, Walter M, Göth C, Oberstein TJ, Linning P, Knölker HJ, Kornhuber J, Maler JM (2020) Pharmacological inhibition of amyloidogenic APP processing and knock-down of APP in primary human macrophages impairs the secretion of cytokines. Front Immunol 11:1967
- Kucheryavykh LY, Kucheryavykh YV, Washington AV, Inyushin MY (2018) Amyloid beta peptide is released during thrombosis in the skin. Int J Mol Sci 19:1705
- Kucheryavykh LY, Dávila-Rodríguez J, Rivera-Aponte DE, Zueva LV, Washington AV, Sanabria P, Inyushin MY (2017) Platelets are responsible for the accumulation of β-amyloid in blood clots inside and around blood vessels in mouse brain after thrombosis. Brain Res Bull 128:98-105
- Tsai PW, Yang CY, Chang HT, Lan CY (2011) Human antimicrobial peptide LL-37 inhibits adhesion of
Candida albicans by interacting with yeast cell-wall carbohydrates. PLoS One 6:e17755
- Kagan BL, Jang H, Capone R, Teran Arce F, Ramachandran S, Lal R, Nussinov R (2012) Antimicrobial properties of amyloid peptides. Mol Pharm 9:708-717
- Inyushin M, Zayas-Santiago A, Rojas L, Kucheryavykh L (2020) On the role of platelet-generated amyloid beta peptides in certain amyloidosis health complications. Front Immunol 11:571083
- Brothers HM, Gosztyla ML, Robinson SR (2018) The physiological roles of amyloid-β peptide hint at new ways to treat Alzheimer's disease. Front Aging Neurosci 10:118
- Morley JE, Farr SA (2014) The role of amyloid-beta in the regulation of memory. Biochem Pharmacol 88:479-485
- Musardo S, Marcello E (2017) Synaptic dysfunction in Alzheimer's disease: from the role of amyloid β-peptide to the α-secretase ADAM10. Eur J Pharmacol 817:30-37
- Lanni C, Fagiani F, Racchi M, Preda S, Pascale A, Grilli M, Allegri N, Govoni S (2018) Beta-amyloid short- and long-term synaptic entanglement. Pharmacol Res 139:243-260
- Puzzo D, Privitera L, Palmeri A (2012) Hormetic effect of amyloid-β peptide in synaptic plasticity and memory. Neurobiol Aging 33:1484.e15-e24
- Cirrito JR, Yamada KA, Finn MB, Sloviter RS, Bales KR, May PC, Schoepp DD, Paul SM, Mennerick S, Holtzman DM (2005) Synaptic activity regulates interstitial fluid amyloid-beta levels in vivo. Neuron 48:913-922
- Cirrito JR, Kang JE, Lee J, Stewart FR, Verges DK, Silverio LM, Bu G, Mennerick S, Holtzman DM (2008) Endocytosis is required for synaptic activity-dependent release of amyloid-beta in vivo. Neuron 58:42-51
- Fagiani F, Lanni C, Racchi M, Pascale A, Govoni S (2019) Amyloid-β and synaptic vesicle dynamics: a cacophonic orchestra. J Alzheimers Dis 72:1-14
- Garcia-Osta A, Alberini CM (2009) Amyloid beta mediates memory formation. Learn Mem 16:267-272
- Morley JE, Farr SA, Banks WA, Johnson SN, Yamada KA, Xu L (2010) A physiological role for amyloid-beta protein: enhancement of learning and memory. J Alzheimers Dis 19:441-449
- Bliss TV, Collingridge GL (1993) A synaptic model of memory: long-term potentiation in the hippocampus. Nature 361:31-39
- Puzzo D, Privitera L, Fa' M, Staniszewski A, Hashimoto G, Aziz F, Sakurai M, Ribe EM, Troy CM, Mercken M, Jung SS, Palmeri A, Arancio O (2011) Endogenous amyloid-β is necessary for hippocampal synaptic plasticity and memory. Ann Neurol 69:819-830
- Flood JF, Roberts E, Sherman MA, Kaplan BE, Morley JE (1994) Topography of a binding site for small amnestic peptides deduced from structure-activity studies: relation to amnestic effect of amyloid beta protein. Proc Natl Acad Sci U S A 91:380-384
- Martinsson I, Capetillo-Zarate E, Faideau M, Willén K, Esteras N, Frykman S, Tjernberg LO, Gouras GK (2019) APP depletion alters selective pre- and post-synaptic proteins. Mol Cell Neurosci 95:86-95
- Young-Pearse TL, Bai J, Chang R, Zheng JB, LoTurco JJ, Selkoe DJ (2007) A critical function for beta-amyloid precursor protein in neuronal migration revealed by in utero RNA interference. J Neurosci 27:14459-14469
- Saura CA, Choi SY, Beglopoulos V, Malkani S, Zhang D, Shankaranarayana Rao BS, Chattarji S, Kelleher RJ 3rd, Kandel ER, Duff K, Kirkwood A, Shen J (2004) Loss of presenilin function causes impairments of memory and synaptic plasticity followed by age-dependent neurodegeneration. Neuron 42:23-36
- Seabrook GR, Smith DW, Bowery BJ, Easter A, Reynolds T, Fitzjohn SM, Morton RA, Zheng H, Dawson GR, Sirinathsinghji DJ, Davies CH, Collingridge GL, Hill RG (1999) Mechanisms contributing to the deficits in hippocampal synaptic plasticity in mice lacking amyloid precursor protein. Neuropharmacology 38:349-359
- Dawson GR, Seabrook GR, Zheng H, Smith DW, Graham S, O'Dowd G, Bowery BJ, Boyce S, Trumbauer ME, Chen HY, Van der Ploeg LH, Sirinathsinghji DJ (1999) Age-related cognitive deficits, impaired long-term potentiation and reduction in synaptic marker density in mice lacking the beta-amyloid precursor protein. Neuroscience 90:1-13
- Laird FM, Cai H, Savonenko AV, Farah MH, He K, Melnikova T, Wen H, Chiang HC, Xu G, Koliatsos VE, Borchelt DR, Price DL, Lee HK, Wong PC (2005) BACE1, a major determinant of selective vulnerability of the brain to amyloid-beta amyloidogenesis, is essential for cognitive, emotional, and synaptic functions. J Neurosci 25:11693-11709
- Vassar R, Bennett BD, Babu-Khan S, Kahn S, Mendiaz EA, Denis P, Teplow DB, Ross S, Amarante P, Loeloff R, Luo Y, Fisher S, Fuller J, Edenson S, Lile J, Jarosinski MA, Biere AL, Curran E, Burgess T, Louis JC, Collins F, Treanor J, Rogers G, Citron M (1999) Beta-secretase cleavage of Alzheimer's amyloid precursor protein by the transmembrane aspartic protease BACE. Science 286:735-741
- De Strooper B, Annaert W, Cupers P, Saftig P, Craessaerts K, Mumm JS, Schroeter EH, Schrijvers V, Wolfe MS, Ray WJ, Goate A, Kopan R (1999) A presenilin-1-dependent gamma-secretase-like protease mediates release of Notch intracellular domain. Nature 398:518-522
- Esch FS, Keim PS, Beattie EC, Blacher RW, Culwell AR, Oltersdorf T, McClure D, Ward PJ (1990) Cleavage of amyloid beta peptide during constitutive processing of its precursor. Science 248:1122-1124
- Pawlik M, Sastre M, Calero M, Mathews PM, Schmidt SD, Nixon RA, Levy E (2004) Overexpression of human cystatin C in transgenic mice does not affect levels of endogenous brain amyloid Beta Peptide. J Mol Neurosci 22:13-18
- Ramsden M, Nyborg AC, Murphy MP, Chang L, Stanczyk FZ, Golde TE, Pike CJ (2003) Androgens modulate beta-amyloid levels in male rat brain. J Neurochem 87:1052-1055
- Cirrito JR, May PC, O'Dell MA, Taylor JW, Parsadanian M, Cramer JW, Audia JE, Nissen JS, Bales KR, Paul SM, DeMattos RB, Holtzman DM (2003) In vivo assessment of brain interstitial fluid with microdialysis reveals plaque-associated changes in amyloid-beta metabolism and half-life. J Neurosci 23:8844-8853
- Giedraitis V, Sundelöf J, Irizarry MC, Gårevik N, Hyman BT, Wahlund LO, Ingelsson M, Lannfelt L (2007) The normal equilibrium between CSF and plasma amyloid beta levels is disrupted in Alzheimer's disease. Neurosci Lett 427:127-131
- Puzzo D, Privitera L, Leznik E, Fà M, Staniszewski A, Palmeri A, Arancio O (2008) Picomolar amyloid-beta positively modulates synaptic plasticity and memory in hippocampus. J Neurosci 28:14537-14545
- Selkoe DJ (2002) Alzheimer's disease is a synaptic failure. Science 298:789-791
- Mattson MP (2008) Hormesis defined. Ageing Res Rev 7:1-7
- Flood JF, Morley JE, Roberts E (1994) An amyloid beta-protein fragment, A beta[12-28], equipotently impairs post-training memory processing when injected into different limbic system structures. Brain Res 663:271-276
- Gulisano W, Melone M, Li Puma DD, Tropea MR, Palmeri A, Arancio O, Grassi C, Conti F, Puzzo D (2018) The effect of amyloid-β peptide on synaptic plasticity and memory is influenced by different isoforms, concentrations, and aggregation status. Neurobiol Aging 71:51-60
- Kayed R, Head E, Thompson JL, McIntire TM, Milton SC, Cotman CW, Glabe CG (2003) Common structure of soluble amyloid oligomers implies common mechanism of pathogenesis. Science 300:486-489
- Iwata N, Mizukami H, Shirotani K, Takaki Y, Muramatsu S, Lu B, Gerard NP, Gerard C, Ozawa K, Saido TC (2004) Presynaptic localization of neprilysin contributes to efficient clearance of amyloid-beta peptide in mouse brain. J Neurosci 24:991-998
- Abramov E, Dolev I, Fogel H, Ciccotosto GD, Ruff E, Slutsky I (2009) Amyloid-beta as a positive endogenous regulator of release probability at hippocampal synapses. Nat Neurosci 12:1567-1576
- Levin ED (2002) Nicotinic receptor subtypes and cognitive function. J Neurobiol 53:633-640
- Albuquerque EX, Pereira EF, Alkondon M, Rogers SW (2009) Mammalian nicotinic acetylcholine receptors: from structure to function. Physiol Rev 89:73-120
- Gray R, Rajan AS, Radcliffe KA, Yakehiro M, Dani JA (1996) Hippocampal synaptic transmission enhanced by low concentrations of nicotine. Nature 383:713-716
- Radcliffe KA, Dani JA (1998) Nicotinic stimulation produces multiple forms of increased glutamatergic synaptic transmission. J Neurosci 18:7075-7083
- Fodero LR, Mok SS, Losic D, Martin LL, Aguilar MI, Barrow CJ, Livett BG, Small DH (2004) Alpha7-nicotinic acetylcholine receptors mediate an Abeta(1-42)-induced increase in the level of acetylcholinesterase in primary cortical neurones. J Neurochem 88:1186-1193
- Dineley KT, Bell KA, Bui D, Sweatt JD (2002) beta-Amyloid peptide activates alpha 7 nicotinic acetylcholine receptors expressed in Xenopus oocytes. J Biol Chem 277:25056-25061
- Sui L, Wang J, Li BM (2008) Role of the phosphoinositide 3-kinase-Akt-mammalian target of the rapamycin signaling pathway in long-term potentiation and trace fear conditioning memory in rat medial prefrontal cortex. Learn Mem 15:762-776
- Kumar A, Lana E, Kumar R, Lithner CU, Darreh-Shori T (2018) Soluble Aβ42 acts as allosteric activator of the core cholinergic enzyme choline acetyltransferase. Front Mol Neurosci 11:327
- Grassi F, Palma E, Tonini R, Amici M, Ballivet M, Eusebi F (2003) Amyloid beta(1-42) peptide alters the gating of human and mouse alpha-bungarotoxin-sensitive nicotinic receptors. J Physiol 547(Pt 1):147-157
- Small DH, Maksel D, Kerr ML, Ng J, Hou X, Chu C, Mehrani H, Unabia S, Azari MF, Loiacono R, Aguilar MI, Chebib M (2007) The beta-amyloid protein of Alzheimer's disease binds to membrane lipids but does not bind to the alpha7 nicotinic acetylcholine receptor. J Neurochem 101:1527-1538
- Dougherty JJ, Wu J, Nichols RA (2003) Beta-amyloid regulation of presynaptic nicotinic receptors in rat hippocampus and neocortex. J Neurosci 23:6740-6747
- Wang HY, Lee DH, D'Andrea MR, Peterson PA, Shank RP, Reitz AB (2002) beta-Amyloid(1-42) binds to alpha7 nicotinic acetylcholine receptor with high affinity. Implications for Alzheimer's disease pathology. J Biol Chem 275:5626-5632
- Snyder EM, Nong Y, Almeida CG, Paul S, Moran T, Choi EY, Nairn AC, Salter MW, Lombroso PJ, Gouras GK, Greengard P (2005) Regulation of NMDA receptor trafficking by amyloid-beta. Nat Neurosci 8:1051-1058
- Almeida CG, Tampellini D, Takahashi RH, Greengard P, Lin MT, Snyder EM, Gouras GK (2005) Beta-amyloid accumulation in APP mutant neurons reduces PSD-95 and GluR1 in synapses. Neurobiol Dis 20:187-198
- Tu S, Okamoto S, Lipton SA, Xu H (2014) Oligomeric Aβ-induced synaptic dysfunction in Alzheimer's disease. Mol Neurodegener 9:48
- Li S, Hong S, Shepardson NE, Walsh DM, Shankar GM, Selkoe D (2009) Soluble oligomers of amyloid Beta protein facilitate hippocampal long-term depression by disrupting neuronal glutamate uptake. Neuron 62:788-801
- Kullmann DM, Lamsa KP (2007) Long-term synaptic plasticity in hippocampal interneurons. Nat Rev Neurosci 8:687-699
- Kamenetz F, Tomita T, Hsieh H, Seabrook G, Borchelt D, Iwatsubo T, Sisodia S, Malinow R (2003) APP processing and synaptic function. Neuron 37:925-937
- Fogel H, Frere S, Segev O, Bharill S, Shapira I, Gazit N, O'Malley T, Slomowitz E, Berdichevsky Y, Walsh DM, Isacoff EY, Hirsch JA, Slutsky I (2014) APP homodimers transduce an amyloid-β-mediated increase in release probability at excitatory synapses. Cell Rep 7:1560-1576
- Giuffrida ML, Tomasello MF, Pandini G, Caraci F, Battaglia G, Busceti C, Di Pietro P, Pappalardo G, Attanasio F, Chiechio S, Bagnoli S, Nacmias B, Sorbi S, Vigneri R, Rizzarelli E, Nicoletti F, Copani A (2015) Monomeric ß-amyloid interacts with type-1 insulin-like growth factor receptors to provide energy supply to neurons. Front Cell Neurosci 9:297
- Giuffrida ML, Caraci F, Pignataro B, Cataldo S, De Bona P, Bruno V, Molinaro G, Pappalardo G, Messina A, Palmigiano A, Garozzo D, Nicoletti F, Rizzarelli E, Copani A (2009) Beta-amyloid monomers are neuroprotective. J Neurosci 29:10582-10587
- Franke TF, Kaplan DR, Cantley LC (1997) PI3K: downstream AKTion blocks apoptosis. Cell 88:435-437
- Alberini CM, Chen DY (2012) Memory enhancement: consolidation, reconsolidation and insulin-like growth factor 2. Trends Neurosci 35:274-283
- Zimbone S, Monaco I, Gianì F, Pandini G, Copani AG, Giuffrida ML, Rizzarelli E (2018) Amyloid Beta monomers regulate cyclic adenosine monophosphate response element binding protein functions by activating type-1 insulin-like growth factor receptors in neuronal cells. Aging Cell 17:e12684
- Nagahara AH, Tuszynski MH (2011) Potential therapeutic uses of BDNF in neurological and psychiatric disorders. Nat Rev Drug Discov 10:209-219
- Fleminger S, Oliver DL, Lovestone S, Rabe-Hesketh S, Giora A (2003) Head injury as a risk factor for Alzheimer's disease: the evidence 10 years on; a partial replication. J Neurol Neurosurg Psychiatry 74:857-862
- Li Y, Li Y, Li X, Zhang S, Zhao J, Zhu X, Tian G (2017) Head injury as a risk factor for dementia and Alzheimer's disease: a systematic review and meta-analysis of 32 observational studies. PLoS One 12:e0169650
- Plassman BL, Havlik RJ, Steffens DC, Helms MJ, Newman TN, Drosdick D, Phillips C, Gau BA, Welsh-Bohmer KA, Burke JR, Guralnik JM, Breitner JC (2000) Documented head injury in early adulthood and risk of Alzheimer's disease and other dementias. Neurology 55:1158-1166
- Guo Z, Cupples LA, Kurz A, Auerbach SH, Volicer L, Chui H, Green RC, Sadovnick AD, Duara R, DeCarli C, Johnson K, Go RC, Growdon JH, Haines JL, Kukull WA, Farrer LA (2000) Head injury and the risk of AD in the MIRAGE study. Neurology 54:1316-1323
- Mayeux R, Ottman R, Tang MX, Noboa-Bauza L, Marder K, Gurland B, Stern Y (1993) Genetic susceptibility and head injury as risk factors for Alzheimer's disease among community-dwelling elderly persons and their first-degree relatives. Ann Neurol 33:494-501
- Johnson VE, Stewart W, Smith DH (2010) Traumatic brain injury and amyloid-β pathology: a link to Alzheimer's disease?. Nat Rev Neurosci 11:361-370
- Tsitsopoulos PP, Marklund N (2013) Amyloid-β peptides and tau protein as biomarkers in cerebrospinal and interstitial fluid following traumatic brain injury: a review of experimental and clinical studies. Front Neurol 4:79
- Scott G, Ramlackhansingh AF, Edison P, Hellyer P, Cole J, Veronese M, Leech R, Greenwood RJ, Turkheimer FE, Gentleman SM, Heckemann RA, Matthews PM, Brooks DJ, Sharp DJ (2016) Amyloid pathology and axonal injury after brain trauma. Neurology 86:821-828
- Chen XH, Johnson VE, Uryu K, Trojanowski JQ, Smith DH (2009) A lack of amyloid beta plaques despite persistent accumulation of amyloid beta in axons of long-term survivors of traumatic brain injury. Brain Pathol 19:214-223
- Loane DJ, Pocivavsek A, Moussa CE, Thompson R, Matsuoka Y, Faden AI, Rebeck GW, Burns MP (2009) Amyloid precursor protein secretases as therapeutic targets for traumatic brain injury. Nat Med 15:377-379
- Pajoohesh-Ganji A, Burns MP, Pal-Ghosh S, Tadvalkar G, Hokenbury NG, Stepp MA, Faden AI (2014) Inhibition of amyloid precursor protein secretases reduces recovery after spinal cord injury. Brain Res 1560:73-82
- Iwata A, Chen XH, McIntosh TK, Browne KD, Smith DH (2002) Long-term accumulation of amyloid-beta in axons following brain trauma without persistent upregulation of amyloid precursor protein genes. J Neuropathol Exp Neurol 61:1056-1068
- Tran HT, Sanchez L, Esparza TJ, Brody DL (2011) Distinct temporal and anatomical distributions of amyloid-β and tau abnormalities following controlled cortical impact in transgenic mice. PLoS One 6:e25475
- Tran HT, LaFerla FM, Holtzman DM, Brody DL (2011) Controlled cortical impact traumatic brain injury in 3xTg-AD mice causes acute intra-axonal amyloid-β accumulation and independently accelerates the development of tau abnormalities. J Neurosci 31:9513-9525
- Abrahamson EE, Ikonomovic MD, Ciallella JR, Hope CE, Paljug WR, Isanski BA, Flood DG, Clark RS, DeKosky ST (2006) Caspase inhibition therapy abolishes brain trauma-induced increases in Abeta peptide: implications for clinical outcome. Exp Neurol 197:437-450
- Washington PM, Morffy N, Parsadanian M, Zapple DN, Burns MP (2014) Experimental traumatic brain injury induces rapid aggregation and oligomerization of amyloid-beta in an Alzheimer's disease mouse model. J Neurotrauma 31:125-134
- Marklund N, Farrokhnia N, Hånell A, Vanmechelen E, Enblad P, Zetterberg H, Blennow K, Hillered L (2014) Monitoring of β-amyloid dynamics after human traumatic brain injury. J Neurotrauma 31:42-55
- Abu Hamdeh S, Waara ER, Möller C, Söderberg L, Basun H, Alafuzoff I, Hillered L, Lannfelt L, Ingelsson M, Marklund N (2018) Rapid amyloid-β oligomer and protofibril accumulation in traumatic brain injury. Brain Pathol 28:451-462
- Olsson A, Csajbok L, Ost M, Höglund K, Nylén K, Rosengren L, Nellgård B, Blennow K (2004) Marked increase of beta-amyloid(1-42) and amyloid precursor protein in ventricular cerebrospinal fluid after severe traumatic brain injury. J Neurol 251:870-876
- Raby CA, Morganti-Kossmann MC, Kossmann T, Stahel PF, Watson MD, Evans LM, Mehta PD, Spiegel K, Kuo YM, Roher AE, Emmerling MR (1998) Traumatic brain injury increases beta-amyloid peptide 1-42 in cerebrospinal fluid. J Neurochem 71:2505-2509
- Emmerling MR, Morganti-Kossmann MC, Kossmann T, Stahel PF, Watson MD, Evans LM, Mehta PD, Spiegel K, Kuo YM, Roher AE, Raby CA (2000) Traumatic brain injury elevates the Alzheimer's amyloid peptide A beta 42 in human CSF. A possible role for nerve cell injury. Ann N Y Acad Sci 903:118-122
- Brody DL, Magnoni S, Schwetye KE, Spinner ML, Esparza TJ, Stocchetti N, Zipfel GJ, Holtzman DM (2008) Amyloid-beta dynamics correlate with neurological status in the injured human brain. Science 321:1221-1224
- Mannix RC, Zhang J, Park J, Lee C, Whalen MJ (2011) Detrimental effect of genetic inhibition of B-site APP-cleaving enzyme 1 on functional outcome after controlled cortical impact in young adult mice. J Neurotrauma 28:1855-1861
- Miners JS, van Helmond Z, Kehoe PG, Love S (2010) Changes with age in the activities of beta-secretase and the Abeta-degrading enzymes neprilysin, insulin-degrading enzyme and angiotensin-converting enzyme. Brain Pathol 20:794-802
- Tokutomi T, Miyagi T, Ogawa T, Ono J, Kawamata T, Sakamoto T, Shigemori M, Nakamura N (2008) Age-associated increases in poor outcomes after traumatic brain injury: a report from the Japan Neurotrauma Data Bank. J Neurotrauma 25:1407-1414
- Mannix RC, Zhang J, Berglass J, Qui J, Whalen MJ (2013) Beneficial effect of amyloid beta after controlled cortical impact. Brain Inj 27:743-748
- Farah MH, Pan BH, Hoffman PN, Ferraris D, Tsukamoto T, Nguyen T, Wong PC, Price DL, Slusher BS, Griffin JW (2011) Reduced BACE1 activity enhances clearance of myelin debris and regeneration of axons in the injured peripheral nervous system. J Neurosci 31:5744-5754
- Tallon C, Rockenstein E, Masliah E, Farah MH (2017) Increased BACE1 activity inhibits peripheral nerve regeneration after injury. Neurobiol Dis 106:147-157
- Tallon C, Marshall KL, Kennedy ME, Hyde LA, Farah MH (2020) Pharmacological BACE inhibition improves axonal regeneration in nerve injury and disease models. neurotherapeutics 17:973-988
- Hu X, He W, Diaconu C, Tang X, Kidd GJ, Macklin WB, Trapp BD, Yan R (2008) Genetic deletion of BACE1 in mice affects remyelination of sciatic nerves. FASEB J 22:2970-2980
- Hu X, Hu J, Dai L, Trapp B, Yan R (2015) Axonal and Schwann cell BACE1 is equally required for remyelination of peripheral nerves. J Neurosci 35:3806-3814
- Baaklini CS, Rawji KS, Duncan GJ, Ho MFS, Plemel JR (2019) Central nervous system remyelination: roles of glia and innate immune cells. Front Mol Neurosci 12:225
- Nave KA, Salzer JL (2006) Axonal regulation of myelination by neuregulin 1. Curr Opin Neurobiol 16:492-500
- Mei L, Xiong WC (2008) Neuregulin 1 in neural development, synaptic plasticity and schizophrenia. Nat Rev Neurosci 9:437-452
- He W, Hu J, Xia Y, Yan R (2014) β-site amyloid precursor protein cleaving enzyme 1(BACE1) regulates Notch signaling by controlling the cleavage of Jagged 1 (Jag1) and Jagged 2 (Jag2) proteins. J Biol Chem 289:20630-20637
- Jendroska K, Poewe W, Daniel SE, Pluess J, Iwerssen-Schmidt H, Paulsen J, Barthel S, Schelosky L, Cervós-Navarro J, DeArmond SJ (1995) Ischemic stress induces deposition of amyloid beta immunoreactivity in human brain. Acta Neuropathol 90:461-466
- Lee PH, Bang OY, Hwang EM, Lee JS, Joo US, Mook-Jung I, Huh K (2005) Circulating beta amyloid protein is elevated in patients with acute ischemic stroke. J Neural Transm (Vienna) 112:1371-1379
- Clarke J, Thornell A, Corbett D, Soininen H, Hiltunen M, Jolkkonen J (2007) Overexpression of APP provides neuroprotection in the absence of functional benefit following middle cerebral artery occlusion in rats. Eur J Neurosci 26:1845-1852
- Grant JL, Ghosn EE, Axtell RC, Herges K, Kuipers HF, Woodling NS, Andreasson K, Herzenberg LA, Herzenberg LA, Steinman L (2012) Reversal of paralysis and reduced inflammation from peripheral administration of β-amyloid in TH1 and TH17 versions of experimental autoimmune encephalomyelitis. Sci Transl Med 4:145ra105
- Maigler KC, Buhr TJ, Park CS, Miller SA, Kozlowski DA, Marr RA (2021) Assessment of the effects of altered amyloid-beta clearance on behavior following repeat closed-head brain injury in amyloid-beta precursor protein humanized mice. J Neurotrauma 38:665-676
- Bird SM, Sohrabi HR, Sutton TA, Weinborn M, Rainey-Smith SR, Brown B, Patterson L, Taddei K, Gupta V, Carruthers M, Lenzo N, Knuckey N, Bucks RS, Verdile G, Martins RN (2016) Cerebral amyloid-β accumulation and deposition following traumatic brain injury--A narrative review and meta-analysis of animal studies. Neurosci Biobehav Rev 64:215-228
- Ueno H, Yamaguchi T, Fukunaga S, Okada Y, Yano Y, Hoshino M, Matsuzaki K (2014) Comparison between the aggregation of human and rodent amyloid β-proteins in GM1 ganglioside clusters. Biochemistry 53:7523-7530
- Götz J, Bodea LG, Goedert M (2018) Rodent models for Alzheimer disease. Nat Rev Neurosci 19:583-598
- Otvos L Jr, Szendrei GI, Lee VM, Mantsch HH (1993) Human and rodent Alzheimer beta-amyloid peptides acquire distinct conformations in membrane-mimicking solvents. Eur J Biochem 211:249-257
- De Gasperi R, Gama Sosa MA, Kim SH, Steele JW, Shaughness MC, Maudlin-Jeronimo E, Hall AA, Dekosky ST, McCarron RM, Nambiar MP, Gandy S, Ahlers ST, Elder GA (2012) Acute blast injury reduces brain abeta in two rodent species. Front Neurol 3:177
- Dickstein DL, Walsh J, Brautigam H, Stockton SD Jr, Gandy S, Hof PR (2010) Role of vascular risk factors and vascular dysfunction in Alzheimer's disease. Mt Sinai J Med 77:82-102
- Biron KE, Dickstein DL, Gopaul R, Jefferies WA (2011) Amyloid triggers extensive cerebral angiogenesis causing blood brain barrier permeability and hypervascularity in Alzheimer's disease. PLoS One 6:e23789
- Jefferies WA, Price KA, Biron KE, Fenninger F, Pfeifer CG, Dickstein DL (2013) Adjusting the compass: new insights into the role of angiogenesis in Alzheimer's disease. Alzheimers Res Ther 5:64
- Carmeliet P (2000) Mechanisms of angiogenesis and arteriogenesis. Nat Med 6:389-395
- Atwood CS, Bowen RL, Smith MA, Perry G (2003) Cerebrovascular requirement for sealant, anti-coagulant and remodeling molecules that allow for the maintenance of vascular integrity and blood supply. Brain Res Brain Res Rev 43:164-178
- Ristori E, Donnini S, Ziche M (2020) New insights into blood-brain barrier maintenance: the homeostatic role of β-amyloid precursor protein in cerebral vasculature. Front Physiol 11:1056
- Atwood CS, Bishop GM, Perry G, Smith MA (2002) Amyloid-beta: a vascular sealant that protects against hemorrhage?. J Neurosci Res 70:356
- Buée L, Hof PR, Delacourte A (1997) Brain microvascular changes in Alzheimer's disease and other dementias. Ann N Y Acad Sci 826:7-24
- Xu X, Wang B, Ren C, Hu J, Greenberg DA, Chen T, Xie L, Jin K (2017) Age-related impairment of vascular structure and functions. Aging Dis 8:590-610
- Kalaria RN, Hedera P (1995) Differential degeneration of the cerebral microvasculature in Alzheimer's disease. Neuroreport 6:477-480
- Mancardi GL, Perdelli F, Rivano C, Leonardi A, Bugiani O (1980) Thickening of the basement membrane of cortical capillaries in Alzheimer's disease. Acta Neuropathol 49:79-83
- Pfeifer LA, White LR, Ross GW, Petrovitch H, Launer LJ (2002) Cerebral amyloid angiopathy and cognitive function: the HAAS autopsy study. Neurology 58:1629-1634
- Balin BJ, Gérard HC, Arking EJ, Appelt DM, Branigan PJ, Abrams JT, Whittum-Hudson JA, Hudson AP (1998) Identification and localization of Chlamydia pneumoniae in the Alzheimer's brain. Med Microbiol Immunol 187:23-42
- MacIntyre A, Hammond CJ, Little CS, Appelt DM, Balin BJ (2002) Chlamydia pneumoniae infection alters the junctional complex proteins of human brain microvascular endothelial cells. FEMS Microbiol Lett 217:167-72
- Desai BS, Schneider JA, Li JL, Carvey PM, Hendey B (2009) Evidence of angiogenic vessels in Alzheimer's disease. J Neural Transm (Vienna) 116:587-597
- Cantara S, Donnini S, Morbidelli L, Giachetti A, Schulz R, Memo M, Ziche M (2004) Physiological levels of amyloid peptides stimulate the angiogenic response through FGF-2. FASEB J 18:1943-1945
- Boscolo E, Folin M, Nico B, Grandi C, Mangieri D, Longo V, Scienza R, Zampieri P, Conconi MT, Parnigotto PP, Ribatti D (2007) Beta amyloid angiogenic activity in vitro and in vivo. Int J Mol Med 19:581-587
- Donnini S, Solito R, Cetti E, Corti F, Giachetti A, Carra S, Beltrame M, Cotelli F, Ziche M (2010) Abeta peptides accelerate the senescence of endothelial cells in vitro and in vivo, impairing angiogenesis. FASEB J 24:2385-2395
- Paris D, Townsend K, Quadros A, Humphrey J, Sun J, Brem S, Wotoczek-Obadia M, DelleDonne A, Patel N, Obregon DF, Crescentini R, Abdullah L, Coppola D, Rojiani AM, Crawford F, Sebti SM, Mullan M (2004) Inhibition of angiogenesis by Abeta peptides. Angiogenesis 7:75-85
- Friesel R, Maciag T (1999) Fibroblast growth factor prototype release and fibroblast growth factor receptor signaling. Thromb Haemost 82:748-754
- Araki S, Shimada Y, Kaji K, Hayashi H (1990) Apoptosis of vascular endothelial cells by fibroblast growth factor deprivation. Biochem Biophys Res Commun 168:1194-1200
- Cameron DJ, Galvin C, Alkam T, Sidhu H, Ellison J, Luna S, Ethell DW (2012) Alzheimer's-related peptide amyloid-β plays a conserved role in angiogenesis. PLoS One 7:e39598
- Cunvong K, Huffmire D, Ethell DW, Cameron DJ (2013) Amyloid-β increases capillary bed density in the adult zebrafish retina. Invest Ophthalmol Vis Sci 54:1516-1521
- Luna S, Cameron DJ, Ethell DW (2013) Amyloid-β and APP deficiencies cause severe cerebrovascular defects: important work for an old villain. PLoS One 8:e75052
- Paris D, Ait-Ghezala G, Mathura VS, Patel N, Quadros A, Laporte V, Mullan M (2005) Anti-angiogenic activity of the mutant Dutch A(beta) peptide on human brain microvascular endothelial cells. Brain Res Mol Brain Res 136:212-230
- Patel NS, Quadros A, Brem S, Wotoczek-Obadia M, Mathura VS, Laporte V, Mullan M, Paris D (2008) Potent anti-angiogenic motifs within the Alzheimer beta-amyloid peptide. Amyloid 15:5-19
- Olofsson A, Sauer-Eriksson AE, Ohman A (2006) The solvent protection of alzheimer amyloid-beta-(1-42) fibrils as determined by solution NMR spectroscopy. J Biol Chem 281:477-483
- Shi HB, Tang B, Liu YW, Wang XF, Chen GJ (2015) Alzheimer disease and cancer risk: a meta-analysis. J Cancer Res Clin Oncol 141:485-494. Erratum in: J Cancer Res Clin Oncol 141: 571.
- Shafi O (2016) Inverse relationship between Alzheimer's disease and cancer, and other factors contributing to Alzheimer's disease: a systematic review. BMC Neurol 16:236
- Ospina-Romero M, Glymour MM, Hayes-Larson E, Mayeda ER, Graff RE, Brenowitz WD, Ackley SF, Witte JS, Kobayashi LC (2020) Association between Alzheimer disease and cancer with evaluation of study biases: a systematic review and meta-analysis. JAMA Netw Open 3:e2025515
- Frain L, Swanson D, Cho K, Gagnon D, Lu KP, Betensky RA, Driver J (2017) Association of cancer and Alzheimer's disease risk in a national cohort of veterans. Alzheimers Dement 13:1364-1370
- Paris D, Ganey N, Banasiak M, Laporte V, Patel N, Mullan M, Murphy SF, Yee GT, Bachmeier C, Ganey C, Beaulieu-Abdelahad D, Mathura VS, Brem S, Mullan M (2010) Impaired orthotopic glioma growth and vascularization in transgenic mouse models of Alzheimer's disease. J Neurosci 30:11251-11258
- Zhao H, Zhu J, Cui K, Xu X, O'Brien M, Wong KK, Kesari S, Xia W, Wong ST (2009) Bioluminescence imaging reveals inhibition of tumor cell proliferation by Alzheimer's amyloid beta protein. Cancer Cell Int 9:15
- Alves da Costa C, Sunyach C, Pardossi-Piquard R, Sévalle J, Vincent B, Boyer N, Kawarai T, Girardot N, St George-Hyslop P, Checler F (2006) Presenilin-dependent gamma-secretase-mediated control of p53-associated cell death in Alzheimer's disease. J Neurosci 26:6377-6385
- Ohyagi Y, Asahara H, Chui DH, Tsuruta Y, Sakae N, Miyoshi K, Yamada T, Kikuchi H, Taniwaki T, Murai H, Ikezoe K, Furuya H, Kawarabayashi T, Shoji M, Checler F, Iwaki T, Makifuchi T, Takeda K, Kira J, Tabira T (2005) Intracellular Abeta42 activates p53 promoter: a pathway to neurodegeneration in Alzheimer's disease. FASEB J 19:255-257
- Chaudhary AK, Yadav N, Bhat TA, O'Malley J, Kumar S, Chandra D (2016) A potential role of X-linked inhibitor of apoptosis protein in mitochondrial membrane permeabilization and its implication in cancer therapy. Drug Discov Today 21:38-47
- Yamamori H, Tanaka T, Kudo T, Takeda M (2004) Amyloid-beta down-regulates XIAP expression in human SH-SY5Y neuroblastoma cells. Neuroreport 15:851-854
- Paradis E, Douillard H, Koutroumanis M, Goodyer C, LeBlanc A (1996) Amyloid beta peptide of Alzheimer's disease downregulates Bcl-2 and upregulates bax expression in human neurons. J Neurosci 16:7533-7539
- Kirkin V, Joos S, Zörnig M (2004) The role of Bcl-2 family members in tumorigenesis. Biochim Biophys Acta 1644:229-249
- Egan MF, Kost J, Tariot PN, Aisen PS, Cummings JL, Vellas B, Sur C, Mukai Y, Voss T, Furtek C, Mahoney E, Harper Mozley L, Vandenberghe R, Mo Y, Michelson D (2018) Randomized trial of verubecestat for mild-to-moderate Alzheimer's disease. N Engl J Med 378:1691-1703
- Sur C, Kost J, Scott D, Adamczuk K, Fox NC, Cummings JL, Tariot PN, Aisen PS, Vellas B, Voss T, Mahoney E, Mukai Y, Kennedy ME, Lines C, Michelson D, Egan MF (2020) BACE inhibition causes rapid, regional, and non-progressive volume reduction in Alzheimer's disease brain. Brain 143:3816-3826
- Novak G, Streffer JR, Timmers M, Henley D, Brashear HR, Bogert J, Russu A, Janssens L, Tesseur I, Tritsmans L, Van Nueten L, Engelborghs S (2020) Long-term safety and tolerability of atabecestat (JNJ-54861911), an oral BACE1 inhibitor, in early Alzheimer's disease spectrum patients: a randomized, double-blind, placebo-controlled study and a two-period extension study. Alzheimers Res Ther 12:58
- Sperling R, Henley D, Aisen PS, Raman R, Donohue MC, Ernstrom K, Rafii MS, Streffer J, Shi Y, Karcher K, Raghavan N, Tymofyeyev Y, Bogert J, Brashear HR, Novak G, Thipphawong J, Saad ZS, Kolb H, Rofael H, Sanga P, Romano G (2021) Findings of efficacy, safety, and biomarker outcomes of atabecestat in preclinical Alzheimer disease: a truncated randomized phase 2b/3 clinical trial. JAMA Neurol 78:293-301
- Wessels AM, Tariot PN, Zimmer JA, Selzler KJ, Bragg SM, Andersen SW, Landry J, Krull JH, Downing AM, Willis BA, Shcherbinin S, Mullen J, Barker P, Schumi J, Shering C, Matthews BR, Stern RA, Vellas B, Cohen S, MacSweeney E, Boada M, Sims JR (2020) Efficacy and safety of lanabecestat for treatment of early and mild Alzheimer disease: the AMARANTH and DAYBREAK-ALZ randomized clinical trials. JAMA Neurol 77:199-209
- Nicolas L, Kammerer KP, Schaible J, Link J, Kleiner O, Borta A, Podhorna J, Scholpp J (2015) P3-283: Pharmacokinetics, pharmacodynamics, and safety of the novel bace inhibitor bi1181181 after oral administration of single ascending doses in healthy subjects. Alzheimer's Dement 11(7S_Part_16):740-741
- Lahiri DK, Maloney B, Long JM, Greig NH (2014) Lessons from a BACE1 inhibitor trial: off-site but not off base. Alzheimers Dement 10(5 Suppl):S411-S419
- Yan R (2016) Stepping closer to treating Alzheimer's disease patients with BACE1 inhibitor drugs. Transl Neurodegener 5:13
- Lo AC, Evans CD, Mancini M, Wang H, Shcherbinin S, Lu M, Natanegara F, Willis BA (2021) Phase II (NAVIGATE-AD study) results of LY3202626 effects on patients with mild Alzheimer's disease dementia. J Alzheimers Dis Rep 5:321-336
- Doody RS, Raman R, Farlow M, Iwatsubo T, Vellas B, Joffe S, Kieburtz K, He F, Sun X, Thomas RG, Aisen PS, Siemers E, Sethuraman G, Mohs R (2013) A phase 3 trial of semagacestat for treatment of Alzheimer's disease. N Engl J Med 369:341-350
- Coric V, van Dyck CH, Salloway S, Andreasen N, Brody M, Richter RW, Soininen H, Thein S, Shiovitz T, Pilcher G, Colby S, Rollin L, Dockens R, Pachai C, Portelius E, Andreasson U, Blennow K, Soares H, Albright C, Feldman HH, Berman RM (2012) Safety and tolerability of the γ-secretase inhibitor avagacestat in a phase 2 study of mild to moderate Alzheimer disease. Arch Neurol 69:1430-1440
- Coric V, Salloway S, van Dyck CH, Dubois B, Andreasen N, Brody M, Curtis C, Soininen H, Thein S, Shiovitz T, Pilcher G, Ferris S, Colby S, Kerselaers W, Dockens R, Soares H, Kaplita S, Luo F, Pachai C, Bracoud L, Mintun M, Grill JD, Marek K, Seibyl J, Cedarbaum JM, Albright C, Feldman HH, Berman RM (2015) Targeting prodromal Alzheimer disease with avagacestat: a randomized clinical trial. JAMA Neurol 72:1324-1333
- Green RC, Schneider LS, Amato DA, Beelen AP, Wilcock G, Swabb EA, Zavitz KH (2009) Effect of tarenflurbil on cognitive decline and activities of daily living in patients with mild Alzheimer disease: a randomized controlled trial. JAMA 302:2557-2564
- Gilman S, Koller M, Black RS, Jenkins L, Griffith SG, Fox NC, Eisner L, Kirby L, Rovira MB, Forette F, Orgogozo JM (2005) Clinical effects of Abeta immunization (AN1792) in patients with AD in an interrupted trial. Neurology 64:1553-1562
- Boche D, Donald J, Love S, Harris S, Neal JW, Holmes C, Nicoll JA (2010) Reduction of aggregated Tau in neuronal processes but not in the cell bodies after Abeta42 immunisation in Alzheimer's disease. Acta Neuropathol 120:13-20
- Schneeberger A, Hendrix S, Mandler M, Ellison N, Bürger V, Brunner M, Frölich L, Mimica N, Hort J, Rainer M, Imarhiagbe D, Kurz A, Peters O, Gertz HJ, Tierney L, Mattner F, Schmidt W, Dubois B (2015) Results from a phase II study to assess the clinical and immunological activity of AFFITOPE®AD02 in patients with early Alzheimer's disease. J Prev Alzheimers Dis 2:103-114
- Pasquier F, Sadowsky C, Holstein A, Leterme Gle P, Peng Y, Jackson N, Fox NC, Ketter N, Liu E, Ryan JM (2016) Two phase 2 multiple ascending-dose studies of vanutide cridificar (ACC-001) and QS-21 adjuvant in mild-to-moderate Alzheimer's disease. J Alzheimers Dis 51:1131-1143
- Vandenberghe R, Riviere ME, Caputo A, Sovago J, Maguire RP, Farlow M, Marotta G, Sanchez-Valle R, Scheltens P, Ryan JM, Graf A (2017) Active Aβ immunotherapy CAD106 in Alzheimer's disease: a phase 2b study. Alzheimers Dement (N Y) 3:10-22
- Ferrero J, Williams L, Stella H, Leitermann K, Mikulskis A, O'Gorman J, Sevigny J (2016) First-in-human, double-blind, placebo-controlled, single-dose escalation study of aducanumab (BIIB037) in mild-to-moderate Alzheimer's disease. Alzheimers Dement (N Y) 2:169-176
- Sevigny J, Chiao P, Bussière T, Weinreb PH, Williams L, Maier M, Dunstan R, Salloway S, Chen T, Ling Y, O'Gorman J, Qian F, Arastu M, Li M, Chollate S, Brennan MS, Quintero-Monzon O, Scannevin RH, Arnold HM, Engber T, Rhodes K, Ferrero J, Hang Y, Mikulskis A, Grimm J, Hock C, Nitsch RM, Sandrock A (2016) The antibody aducanumab reduces Aβ plaques in Alzheimer's disease. Nature 537:50-56
- Salloway S, Sperling R, Fox NC, Blennow K, Klunk W, Raskind M, Sabbagh M, Honig LS, Porsteinsson AP, Ferris S, Reichert M, Ketter N, Nejadnik B, Guenzler V, Miloslavsky M, Wang D, Lu Y, Lull J, Tudor IC, Liu E, Grundman M, Yuen E, Black R, Brashear HR (2014) Two phase 3 trials of bapineuzumab in mild-to-moderate Alzheimer's disease. N Engl J Med 370:322-333
- Vandenberghe R, Rinne JO, Boada M, Katayama S, Scheltens P, Vellas B, Tuchman M, Gass A, Fiebach JB, Hill D, Lobello K, Li D, McRae T, Lucas P, Evans I, Booth K, Luscan G, Wyman BT, Hua L, Yang L, Brashear HR, Black RS (2016) Bapineuzumab for mild to moderate Alzheimer's disease in two global, randomized, phase 3 trials. Alzheimers Res Ther 8:18
- Abushouk AI, Elmaraezy A, Aglan A, Salama R, Fouda S, Fouda R, AlSafadi AM (2017) Bapineuzumab for mild to moderate Alzheimer's disease: a meta-analysis of randomized controlled trials. BMC Neurol 17:66
- Delnomdedieu M, Duvvuri S, Li DJ, Atassi N, Lu M, Brashear HR, Liu E, Ness S, Kupiec JW (2016) First-In-Human safety and long-term exposure data for AAB-003 (PF-05236812) and biomarkers after intravenous infusions of escalating doses in patients with mild to moderate Alzheimer's disease. Alzheimers Res Ther 8:12
- Ostrowitzki S, Lasser RA, Dorflinger E, Scheltens P, Barkhof F, Nikolcheva T, Ashford E, Retout S, Hofmann C, Delmar P, Klein G, Andjelkovic M, Dubois B, Boada M, Blennow K, Santarelli L, Fontoura P (2017) A phase III randomized trial of gantenerumab in prodromal Alzheimer's disease. Alzheimers Res Ther 9:95
- Doody RS, Thomas RG, Farlow M, Iwatsubo T, Vellas B, Joffe S, Kieburtz K, Raman R, Sun X, Aisen PS, Siemers E, Liu-Seifert H, Mohs R (2014) Phase 3 trials of solanezumab for mild-to-moderate Alzheimer's disease. N Engl J Med 370:311-321
- Siemers ER, Sundell KL, Carlson C, Case M, Sethuraman G, Liu-Seifert H, Dowsett SA, Pontecorvo MJ, Dean RA, Demattos R (2016) Phase 3 solanezumab trials: secondary outcomes in mild Alzheimer's disease patients. Alzheimers Dement 12:110-120
- Yang T, Dang Y, Ostaszewski B, Mengel D, Steffen V, Rabe C, Bittner T, Walsh DM, Selkoe DJ (2019) Target engagement in an alzheimer trial: Crenezumab lowers amyloid β oligomers in cerebrospinal fluid. Ann Neurol 86:215-224
- Landen JW, Cohen S, Billing CB Jr, Cronenberger C, Styren S, Burstein AH, Sattler C, Lee JH, Jack CR Jr, Kantarci K, Schwartz PF, Duggan WT, Zhao Q, Sprenger K, Bednar MM, Binneman B (2017) Multiple-dose ponezumab for mild-to-moderate Alzheimer's disease: safety and efficacy. Alzheimers Dement (N Y) 3:339-347
- Relkin NR, Thomas RG, Rissman RA, Brewer JB, Rafii MS, van Dyck CH, Jack CR, Sano M, Knopman DS, Raman R, Szabo P, Gelmont DM, Fritsch S, Aisen PS; Alzheimer's Disease Cooperative Study (2017) A phase 3 trial of IV immunoglobulin for Alzheimer disease. Neurology 88:1768-1775
- Lowe SL, Duggan Evans C, Shcherbinin S, Cheng YJ, Willis BA, Gueorguieva I, Lo AC, Fleisher AS, Dage JL, Ardayfio P, Aguiar G, Ishibai M, Takaichi G, Chua L, Mullins G, Sims JR (2021) Donanemab (LY3002813) phase 1b study in Alzheimer's disease: rapid and sustained reduction of brain amyloid measured by florbetapir F18 imaging. J Prev Alzheimers Dis 8:414-424
- Ayton S (2021) Brain volume loss due to donanemab. Eur J Neurol 28:e67-e68
- Swanson CJ, Zhang Y, Dhadda S, Wang J, Kaplow J, Lai RYK, Lannfelt L, Bradley H, Rabe M, Koyama A, Reyderman L, Berry DA, Berry S, Gordon R, Kramer LD, Cummings JL (2021) A randomized, double-blind, phase 2b proof-of-concept clinical trial in early Alzheimer's disease with lecanemab, an anti-Aβ protofibril antibody. Alzheimers Res Ther 13:80
- Pradier L, Blanchard-Brégeon V, Bohme A, Debeir T, Menager J, Benoit P, Barneoud P, Taupin V, Bertrand P, Dugay P, Cameron B, Shi Y, Naimi S, Duchesne M, Gagnaire M, Weeden T, Travaline T, Reczek D, Khiroug L, Slaoui M, Brunel P, Fukuyama H, Ravetch J, Canton T, Cohen C (2018) SAR228810: an antibody for protofibrillar amyloid β peptide designed to reduce the risk of amyloid-related imaging abnormalities (ARIA). Alzheimers Res Ther 10:117
- Leyhe T, Andreasen N, Simeoni M, Reich A, von Arnim CA, Tong X, Yeo A, Khan S, Loercher A, Chalker M, Hottenstein C, Zetterberg H, Hilpert J, Mistry P (2014) Modulation of β-amyloid by a single dose of GSK933776 in patients with mild Alzheimer's disease: a phase I study. Alzheimers Res Ther 6:19
- Ritchie CW, Bush AI, Mackinnon A, Macfarlane S, Mastwyk M, MacGregor L, Kiers L, Cherny R, Li QX, Tammer A, Carrington D, Mavros C, Volitakis I, Xilinas M, Ames D, Davis S, Beyreuther K, Tanzi RE, Masters CL (2003) Metal-protein attenuation with iodochlorhydroxyquin (clioquinol) targeting Abeta amyloid deposition and toxicity in Alzheimer disease: a pilot phase 2 clinical trial. Arch Neurol 60:1685-1691
- Wang CY, Wang PN, Chiu MJ, Finstad CL, Lin F, Lynn S, Tai YH, De Fang X, Zhao K, Hung CH, Tseng Y, Peng WJ, Wang J, Yu CC, Kuo BS, Frohna PA (2017) UB-311, a novel UBITh®amyloid β peptide vaccine for mild Alzheimer's disease. Alzheimers Dement (N Y) 3:262-272
- Lacosta AM, Pascual-Lucas M, Pesini P, Casabona D, Pérez-Grijalba V, Marcos-Campos I, Sarasa L, Canudas J, Badi H, Monleón I, San-José I, Munuera J, Rodríguez-Gómez O, Abdelnour C, Lafuente A, Buendía M, Boada M, Tárraga L, Ruiz A, Sarasa M (2018) Safety, tolerability and immunogenicity of an active anti-Aβ40 vaccine (ABvac40) in patients with Alzheimer's disease: a randomised, double-blind, placebo-controlled, phase I trial. Alzheimers Res Ther 10:12
- Aisen PS, Saumier D, Briand R, Laurin J, Gervais F, Tremblay P, Garceau D (2006) A phase II study targeting amyloid-beta with 3APS in mild-to-moderate Alzheimer disease. Neurology 67:1757-1763
- Gauthier S, Aisen PS, Ferris SH, Saumier D, Duong A, Haine D, Garceau D, Suhy J, Oh J, Lau W, Sampalis J (2009) Effect of tramiprosate in patients with mild-to-moderate Alzheimer's disease: exploratory analyses of the MRI sub-group of the Alphase study. J Nutr Health Aging 13:550-557
- Salloway S, Sperling R, Keren R, Porsteinsson AP, van Dyck CH, Tariot PN, Gilman S, Arnold D, Abushakra S, Hernandez C, Crans G, Liang E, Quinn G, Bairu M, Pastrak A, Cedarbaum JM; ELND005-AD201 Investigators (2011) A phase 2 randomized trial of ELND005, scyllo-inositol, in mild to moderate Alzheimer disease. Neurology 77:1253-1262
- Sampson EL, Jenagaratnam L, McShane R (2014) Metal protein attenuating compounds for the treatment of Alzheimer's dementia. Cochrane Database Syst Rev 2:CD005380
- Lannfelt L, Blennow K, Zetterberg H, Batsman S, Ames D, Harrison J, Masters CL, Targum S, Bush AI, Murdoch R, Wilson J, Ritchie CW; PBT2-201-EURO Study Group (2008) Safety, efficacy, and biomarker findings of PBT2 in targeting Abeta as a modifying therapy for Alzheimer's disease: a phase IIa, double-blind, randomised, placebo-controlled trial. Lancet Neurol 7:779-786
- Xiao S, Chan P, Wang T, Hong Z, Wang S, Kuang W, He J, Pan X, Zhou Y, Ji Y, Wang L, Cheng Y, Peng Y, Ye Q, Wang X, Wu Y, Qu Q, Chen S, Li S, Chen W, Xu J, Peng D, Zhao Z, Li Y, Zhang J, Du Y, Chen W, Fan D, Yan Y, Liu X, Zhang W, Luo B, Wu W, Shen L, Liu C, Mao P, Wang Q, Zhao Q, Guo Q, Zhou Y, Li Y, Jiang L, Ren W, Ouyang Y, Wang Y, Liu S, Jia J, Zhang N, Liu Z, He R, Feng T, Lu W, Tang H, Gao P, Zhang Y, Chen L, Wang L, Yin Y, Xu Q, Xiao J, Cong L, Cheng X, Zhang H, Gao D, Xia M, Lian T, Peng G, Zhang X, Jiao B, Hu H, Chen X, Guan Y, Cui R, Huang Q, Xin X, Chen H, Ding Y, Zhang J, Feng T, Cantillon M, Chen K, Cummings JL, Ding J, Geng M, Zhang Z (2021) A 36-week multicenter, randomized, double-blind, placebo-controlled, parallel-group, phase 3 clinical trial of sodium oligomannate for mild-to-moderate Alzheimer's dementia. Alzheimers Res Ther 13:62
- Glynn-Servedio BE, Ranola TS (2017) AChE inhibitors and NMDA receptor antagonists in advanced Alzheimer's disease. Consult Pharm 32:511-518
- Henley D, Raghavan N, Sperling R, Aisen P, Raman R, Romano G (2019) Preliminary results of a trial of atabecestat in preclinical Alzheimer's disease. N Engl J Med 380:1483-1485
- Penninkilampi R, Brothers HM, Eslick GD (2016) Pharmacological agents targeting γ-secretase increase risk of cancer and cognitive decline in Alzheimer's disease patients: a systematic review and meta-analysis. J Alzheimers Dis 53:1395-1404
- Lannfelt L, Relkin NR, Siemers ER (2014) Amyloid-ß-directed immunotherapy for Alzheimer's disease. J Intern Med 275:284-295
- Nicoll JA, Barton E, Boche D, Neal JW, Ferrer I, Thompson P, Vlachouli C, Wilkinson D, Bayer A, Games D, Seubert P, Schenk D, Holmes C (2006) Abeta species removal after abeta42 immunization. J Neuropathol Exp Neurol 65:1040-1048
- Orgogozo JM, Gilman S, Dartigues JF, Laurent B, Puel M, Kirby LC, Jouanny P, Dubois B, Eisner L, Flitman S, Michel BF, Boada M, Frank A, Hock C (2003) Subacute meningoencephalitis in a subset of patients with AD after Abeta42 immunization. Neurology 61:46-54
- Mahase E (2021) Three FDA advisory panel members resign over approval of Alzheimer's drug. BMJ 373:n1503
- Alexander GC, Emerson S, Kesselheim AS (2021) Evaluation of aducanumab for Alzheimer disease: scientific evidence and regulatory review involving efficacy, safety, and futility. JAMA 325:1717-1718
- Knopman DS, Jones DT, Greicius MD (2021) Failure to demonstrate efficacy of aducanumab: an analysis of the EMERGE and ENGAGE trials as reported by Biogen, December 2019. Alzheimers Dement 17:696-701
- Abi-Saab D, Andjelkovic M, Delmar P, Voyle N, Esau N, Lasser RA (2017) [P1-045]: The effect of 6 months' dosing on the rate of amyloid-related imaging abnormalities (aria) in the marguerite road study. Alzheimers Dement 13(7 Suppl Pt 5):P252-P253
- DiFrancesco JC, Longoni M, Piazza F (2015) Anti-Aβ autoantibodies in amyloid related imaging abnormalities (ARIA): candidate biomarker for immunotherapy in Alzheimer's disease and cerebral amyloid angiopathy. Front Neurol 6:207
- Sperling RA, Jack CR Jr, Black SE, Frosch MP, Greenberg SM, Hyman BT, Scheltens P, Carrillo MC, Thies W, Bednar MM, Black RS, Brashear HR, Grundman M, Siemers ER, Feldman HH, Schindler RJ (2011) Amyloid-related imaging abnormalities in amyloid-modifying therapeutic trials: recommendations from the Alzheimer's Association Research Roundtable Workgroup. Alzheimers Dement 7:367-385
- Boche D, Zotova E, Weller RO, Love S, Neal JW, Pickering RM, Wilkinson D, Holmes C, Nicoll JA (2008) Consequence of Abeta immunization on the vasculature of human Alzheimer's disease brain. Brain 131(Pt 12):3299-3310
- Barakos J, Sperling R, Salloway S, Jack C, Gass A, Fiebach JB, Tampieri D, Melançon D, Miaux Y, Rippon G, Black R, Lu Y, Brashear HR, Arrighi HM, Morris KA, Grundman M (2013) MR imaging features of amyloid-related imaging abnormalities. AJNR Am J Neuroradiol 34:1958-1965
- Selkoe DJ (2019) Alzheimer disease and aducanumab: adjusting our approach. Nat Rev Neurol 15:365-366
- Sakono M, Zako T (2010) Amyloid oligomers: formation and toxicity of Abeta oligomers. FEBS J 277:1348-1358
- Ackley SF, Zimmerman SC, Brenowitz WD, Tchetgen Tchetgen EJ, Gold AL, Manly JJ, Mayeda ER, Filshtein TJ, Power MC, Elahi FM, Brickman AM, Glymour MM (2021) Effect of reductions in amyloid levels on cognitive change in randomized trials: instrumental variable meta-analysis. BMJ 372:n156
- Schilling S, Rahfeld JU, Lues I, Lemere CA (2018) Passive Aβ immunotherapy: current achievements and future perspectives. Molecules 23:1068