Articles

Article Tools
 View Full Text View Full Text |
 Abstract Abstract |
 Article as PDF Article as PDF |
 Print this Article Print this Article |
 Pubmed Pubmed |
 PMC PMC |
 PubReader PubReader |
 Export to Citation Export to Citation |
 Email Alerts Email Alerts |
 Open Access Open Access |
Share this article on :



Stats or Metrics
Article
Review Article
Exp Neurobiol 2016; 25(4): 147-155
Published online August 31, 2016
https://doi.org/10.5607/en.2016.25.4.147
© The Korean Society for Brain and Neural Sciences
Prothrombin Kringle-2: A Potential Inflammatory Pathogen in the Parkinsonian Dopaminergic System
Eunju Leem1,2#, Kyoung Hoon Jeong1,2#, So-Yoon Won4, Won-Ho Shin5* and Sang Ryong Kim1,2,3,6*
1School of Life Sciences & Biotechnology, 2BK21 plus KNU Creative BioResearch Group, 3Institute of Life Science & Biotechnology, Kyungpook National University, Daegu 41566, 4Department of Biochemistry and Signaling Disorder Research Center, College of Medicine, Chungbuk National University, Cheongju 28644, 5Predictive Research Center, Korea Institute of Toxicology, Daejeon 34114, 6Brain Science and Engineering Institute, Kyungpook National University, Daegu 41944, Korea
Correspondence to: *To whom correspondence should be addressed.
Sang Ryong Kim, TEL: 82-53-950-7362, FAX: 82-53-943-2762
e-mail: srk75@knu.ac.kr
Won-Ho Shin, TEL: 82-42-610-8088, FAX: 82-42-610-8157
e-mail: whshin@kitox.re.kr
#These authors contributed equally to this work.
Abstract
- Go to
- Abstract
- INTRODUCTION
- Parkinson's disease and microglial activation
- Microglia and neuroinflammation
- Control of neuroin flammation by micro glial TLR4
- Production of pKr-2 and its general roles
in vivo - pKr-2 as an endogenous pathogen in the nigrostriatal DA system
- Control of pKr-2-induced neurotoxicity
- Conclusion
- Figure
- Reference
Although accumulating evidence suggests that microglia-mediated neuroinflammation may be crucial for the initiation and progression of Parkinson's disease (PD), and that the control of neuroinflammation may be a useful strategy for preventing the degeneration of nigrostriatal dopaminergic (DA) projections in the adult brain, it is still unclear what kinds of endogenous biomolecules initiate microglial activation, consequently resulting in neurodegeneration. Recently, we reported that the increase in the levels of prothrombin kringle-2 (pKr-2), which is a domain of prothrombin that is generated by active thrombin, can lead to disruption of the nigrostriatal DA projection. This disruption is mediated by neurotoxic inflammatory events
Keywords: Prothrombin kringle-2, Parkinson’s disease, Microglia, Toll-like receptor 4
INTRODUCTION
- Go to
- Abstract
- INTRODUCTION
- Parkinson's disease and microglial activation
- Microglia and neuroinflammation
- Control of neuroin flammation by micro glial TLR4
- Production of pKr-2 and its general roles
in vivo - pKr-2 as an endogenous pathogen in the nigrostriatal DA system
- Control of pKr-2-induced neurotoxicity
- Conclusion
- Figure
- Reference
Parkinson's disease (PD) is the second-most common neurodegenerative disorder and is characterized by the progressive degeneration of dopaminergic (DA) neurons and a decrease in striatal dopamine. PD is associated with clinical movement disorders, including a tremor at rest, rigidity of the limbs, bradykinesia (slowness and paucity of voluntary movement), and postural instability (a tendency to fall even in the absence of weakness or cerebellar balance disturbance) [1,2,3]. Although we do not fully understand the etiology of PD, accumulating evidence suggests that microglia, which are the resident immune cells of the brain, are crucial mediators of the brain inflammatory processes that lead to neurotoxicity, and that excessive microglial activation contributes to the initiation and progression of PD [3,4,5]. However, it is largely unknown what endogenous biomolecules initiate and stimulate microglial activation, even though the control of microglial activators, which stimulate neurotoxic inflammation, may be a useful strategy for the prevention of the degeneration of the nigrostriatal DA projection in the adult brain.
Toll-like receptors (TLRs) are pattern recognition receptors that recognize specific pathogen-associated molecular signatures and subsequently initiate inflammatory and immune responses [3,6]. TLR4 recognizes various ligands, such as lipopolysaccharide (LPS), envelope proteins, heat-shock proteins, fibrinogen, and hyaluronan [3,6]. The activation of TLR4 in immune cells induces increases in the levels of inflammatory cytokines [3,7]. Although the pattern of TLR expression in the brain is controversial, there are many reports suggesting that microglia are important cells for TLR4-mediated immune responses, which may be involved in neurodegenerative diseases such as Alzheimer's disease (AD) and PD [8,9,10]. Moreover, increases in TLR4 expression have been observed in α-synuclein-overexpressing transgenic mice and in patients with multiple system atrophy [11], although alterations in TLR4 expression in patients with PD are still unclear. These results suggest that an increase in microglial TLR4 may be crucial for the pathogenesis of PD, and that the discovery of endogenous molecules involved in the induction of microglial TLR4 may be useful in guiding the development of knowledge-based targeted therapeutics for PD.
We previously reported that prothrombin kringle-2 (pKr-2), which is a domain of prothrombin that is generated by active thrombin, is able to induce the death of DA neurons in the rat SN through microglial activation, even though pKr-2 itself was not directly toxic to neurons [5]. Moreover, we recently found that patients with PD have increased pKr-2 expression in the SN, and that nigrostriatal DA projections might degenerate due to neurotoxic inflammation following pKr-2 upregulation-induced production of microglial TLR4 in the SN of adult murine brain [3]. These results suggest that pKr-2 might be a potential pathogenic factor in PD, and that limiting pKr-2-induced microglial activation may be an effective therapeutic strategy for protecting DA neurons in the adult brain.
Parkinson's disease and microglial activation
- Go to
- Abstract
- INTRODUCTION
- Parkinson's disease and microglial activation
- Microglia and neuroinflammation
- Control of neuroin flammation by micro glial TLR4
- Production of pKr-2 and its general roles
in vivo - pKr-2 as an endogenous pathogen in the nigrostriatal DA system
- Control of pKr-2-induced neurotoxicity
- Conclusion
- Figure
- Reference
The histopathological features of PD are the idiopathic degeneration of DA neurons in the pars compacta of the SN and loss of DA nerve terminals in the striatum [12]. This progressive neurodegeneration, which consequently results in the reduction of dopamine in the nigrostriatal DA system [12], is generally accompanied by both motor and non-motor symptoms. The non-motor symptoms of PD include olfactory dysfunction, cognitive impairment, psychiatric symptoms, sleep disorders, pain, depression, and rapid eye movement sleep behavior disorders [13]. The motor symptoms of PD include movement disorders such as resting tremor, muscular rigidity, bradykinesia, akinesia, and postural instability [13]. Although the maintenance of the dopamine concentration is considered to be a useful target in the development of therapeutics against PD progression, clinical trials focusing on dopamine production have not been successful [14,15]. Levodopa (L-3,4-dihydroxyphenylalanine), which is a precursor of dopamine, is one of the main drugs used to treat PD symptoms. However, the long-term use of levodopa is associated with complications, such as abnormal involuntary movements called dyskinesias and dystonias [14,15]. Moreover, no treatment has been identified that forestalls deterioration attributable to progressive neurodegeneration [1,2]. These findings indicate that sustained dopamine supplementation alone is unable to protect or restore DA systems during PD progression. Thus, the control of PD pathogenesis using approaches such as inhibition of mitochondrial dysfunction and reduction of activated microglia-derived oxidative stress and/or neuroinflammation, may be more important in treating PD progression than the maintenance of dopamine production [16,17,18,19], even though this is the major therapeutic strategy currently used to treat patients with PD.
Accumulating evidence suggests that microglial activation, which is an important neurotoxic mechanism, plays important roles in the initiation and progression of PD [20,21]. Imamura et al. have previously reported that the accumulation of CR3/43-positive cells (activated microglia) is increased in the SN and putamen in the post-mortem brains of patients with PD [20]. Furthermore, [11C](R)-PK11195, which is a radiotracer used to detect activated microglia, noticeably accumulates in the brains of patients diagnosed with PD [21]. These findings strongly suggest that microglial activation negatively affects neuronal cell survival and consequently leads to neurodegeneration in PD.
Microglia and neuroinflammation
- Go to
- Abstract
- INTRODUCTION
- Parkinson's disease and microglial activation
- Microglia and neuroinflammation
- Control of neuroin flammation by micro glial TLR4
- Production of pKr-2 and its general roles
in vivo - pKr-2 as an endogenous pathogen in the nigrostriatal DA system
- Control of pKr-2-induced neurotoxicity
- Conclusion
- Figure
- Reference
Microglia are the resident immune cells in the central nervous system (CNS). In the resting state, microglia have small cell bodies and numerous processes and can support neuronal function and survival [22]. In pathological conditions, microglia stimulated by various activators undergo phagocytic morphological changes, which are characterized by enlarged cell bodies and short processes, and these altered microglia exert beneficial effects that repair tissue by releasing anti-inflammatory cytokines and neurotrophic factors [22,23]. However, the major cause of neurotoxic inflammation in the CNS is the response of microglia to a variety of stimuli, such as infection, trauma, and toxins [19], and activated microglia produce neurotoxic inflammatory cytokines, such as interleukin (IL)-1β, tumor necrosis factor (TNF)-α, and IL-6 [22,24]. In addition, activated microglia can produce reactive oxygen species (ROS), such as O2- and O2--derived oxidants,
Consistent with the observation of increases in NADPH oxidase and neurotoxic cytokines in the brains of patients with PD [3,19,20,28], there are many reports indicating the significance of these factors involved in the degeneration of the nigrostriatal DA system in animal models of PD. 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) was discovered to be a neurotoxic compound in drug addicts who showed parkinsonian symptoms [29] and has been widely used in the production of PD animal models [30]. In the SN of adult mice, MPTP induces microglial activation, which is involved in neurotoxicity [31]. The blockade of microglial activation following treatment with minocycline, a broad-spectrum tetracycline antibiotic, inhibits the production of microglial-derived deleterious factors, and exerts neuroprotective effects in the MPTP-lesioned SN [30]. These suggest that microglial activation is involved in the PD pathogenesis related to DA neuronal damage. In addition, the increased expression of gp91phox, which is an NADPH oxidase subunit, is observed in the activated microglia of MPTP-injected mice [28]. In fact, the upregulation of gp91phox within activated microglia is markedly reduced by minocycline treatment, which leads to the protection of DA neurons
6-hydroxydopamine (6-OHDA) is also often used to produce PD models for the testing symptomatic therapies and the study of the mechanisms of DA neuronal death [1]. Since the structure of 6-OHDA is similar to that of dopamine, it is taken up into DA neurons by the dopamine transporter, where it acts as a neuronal toxin by generating ROS [30]. In addition to direct neuronal toxicity, 6-OHDA-induced neurotoxicity can promote microglial activation in nigrostriatal DA projection areas [32,33], resulting in the production of neurotoxic proinflammatory cytokines [34,35]. In addition, microglial activation induced by 6-OHDA treatment parallels the activation of NADPH oxidase, including the p47phox and gp91phox subunits, in nigral microglia [36]. Similar to the effects of MPTP, the 6-OHDA-induced toxic effects in DA neurons are attenuated by minocycline treatment [37]. Taken together, these results suggest that microglial-derived neuroinflammation, which includes the production of neurotoxic cytokines and activation of NADPH oxidase within microglia, may play a crucial role in DA neuronal degeneration in animal models of PD. In addition to MPTP and 6-OHDA, studies using rotenone, which is an odorless, colorless, crystalline isoflavone used as a broad-spectrum insecticide, piscicide, and pesticide, have also indicated that the production of neurotoxic cytokines and activation of NADPH oxidase in microglia play important roles in PD pathogenesis [38,39].
The above observations indicate that excessive increases in proinflammatory mediators and ROS production following microglial activation lead to severe neurotoxicity, resulting in the deterioration of the DA system in the adult brain. While numerous studies have examined the development of therapeutic and preventive agents against PD, there are currently no cures that stop or slow down the progressive degeneration of DA neurons and PD symptoms. Therefore, efforts to develop therapeutic and preventive agents for PD need to aim at inhibiting microglial-derived neuroinflammation, which would suppress microglial activation and its pathogenic mechanisms.
Control of neuroin flammation by micro glial TLR4
- Go to
- Abstract
- INTRODUCTION
- Parkinson's disease and microglial activation
- Microglia and neuroinflammation
- Control of neuroin flammation by micro glial TLR4
- Production of pKr-2 and its general roles
in vivo - pKr-2 as an endogenous pathogen in the nigrostriatal DA system
- Control of pKr-2-induced neurotoxicity
- Conclusion
- Figure
- Reference
TLR4 initiates the activation of the innate immune response by recognizing pathogens as a pattern-recognition receptor. LPS is a representative ligand for TLR4 [40]. The TLR4-mediated activation of NF-κB and mitogen-activated protein kinases that occurs
Production of pKr-2 and its general roles in vivo
- Go to
- Abstract
- INTRODUCTION
- Parkinson's disease and microglial activation
- Microglia and neuroinflammation
- Control of neuroin flammation by micro glial TLR4
- Production of pKr-2 and its general roles
in vivo - pKr-2 as an endogenous pathogen in the nigrostriatal DA system
- Control of pKr-2-induced neurotoxicity
- Conclusion
- Figure
- Reference
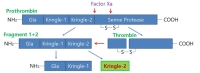
Prothrombin, which is known to be synthesized mainly in the liver and then secreted into the bloodstream [52], is cleaved to produce fragment 1-2 (kringle regions) and thrombin during activation [53,54]. Activated thrombin, a serine protease which converts soluble fibrinogen into insoluble fibrin for blood coagulation, can cleave pKr-1 and -2 (Fig. 1) [55]. The functions of pKr-2 are well-established in the field of angiogenesis. For instance, pKr-2, which is purified from LPS-treated rabbit serum, acts as an angiogenic inhibitor during bovine capillary endothelial cell proliferation [56]. Treatment with pKr-2 induces the suppression of basic fibroblast growth factor-triggered endothelial cell growth and angiogenesis in the chorioallantoic membrane of chick embryos [57]. Moreover, treatment with pKr-2 inhibits endothelial cell proliferation and angiogenesis by inactivating the cyclin D1/cyclin-dependent kinase 4 (CDK4) complex through the induction of ROS production and upregulation of nuclear CDK inhibitors [58]. It also inhibits fibrin formation and platelet aggregation by binding to thrombin and inducing conformational changes at its active site, resulting in a reduction in the clotting activity of thrombin [59]. In addition, recombinant human pKr-2 reduces the immunoreactivity of matrix metalloproteinases 2 and 9 and the expression of vascular endothelial growth factor, which results in the inhibition of B16F10 melanoma cell metastasis [60].
Although many studies have investigated the functions of pKr-2 [56,57,58,59,60] and prothrombin is expressed in brain tissues [52], few reports have examined the roles of pKr-2 in the CNS. The accumulation of prothrombin and thrombin, which might be due to blood-brain barrier leakage, has been shown in the brains of patients with PD and AD [61,62,63], which suggests a possible increase in pKr-2 expression. Moreover, we previously reported that the upregulation of pKr-2 could contribute to microglial activation, resulting in neurodegeneration in the SN of murine brains [3]. Therefore, these results suggest that pKr-2 expression is increased in the lesioned brain and that its upregulation is involved in neurotoxic effects in the adult brain.
pKr-2 as an endogenous pathogen in the nigrostriatal DA system
- Go to
- Abstract
- INTRODUCTION
- Parkinson's disease and microglial activation
- Microglia and neuroinflammation
- Control of neuroin flammation by micro glial TLR4
- Production of pKr-2 and its general roles
in vivo - pKr-2 as an endogenous pathogen in the nigrostriatal DA system
- Control of pKr-2-induced neurotoxicity
- Conclusion
- Figure
- Reference
pKr-2 can trigger microglial activation, resulting in the production of neurotoxic cytokines such as TNF-α and IL-1β, and inflammatory mediators such as inducible nitric oxide synthase (iNOS) and cyclooxygenase-2 [5,64,65]. In addition, pKr-2-activated microglia produce O2- and O2--derived oxidants through the activation of NADPH oxidase, which is also involved in neurotoxic events in the murine cortex [64]. Moreover, we recently suggested that pKr-2 upregulation is involved in the pathogenesis of PD, and that the control of microglial pKr-2 expression and pKr-2-induced microglial TLR4 overexpression might be important for protecting the nigrostriatal DA system against PD [3]. These results help the understanding of the relationships among pKr-2, microglia, and TLR4 (Fig. 2). To ascertain whether pKr-2 is involved in PD
Control of pKr-2-induced neurotoxicity
- Go to
- Abstract
- INTRODUCTION
- Parkinson's disease and microglial activation
- Microglia and neuroinflammation
- Control of neuroin flammation by micro glial TLR4
- Production of pKr-2 and its general roles
in vivo - pKr-2 as an endogenous pathogen in the nigrostriatal DA system
- Control of pKr-2-induced neurotoxicity
- Conclusion
- Figure
- Reference
Since inhibiting inflammation is an important strategy for the prevention of neuronal damage in neurodegenerative disease, therapies for the control of the activation of microglia have been proposed. Although the exact mechanisms are unknown, our recent results strongly suggest that TLR4 and pKr-2 are closely associated with neuroinflammation in PD [3]. Similar to our results involved in neurodegeneration in the DA system, previous studies have shown that TLR4 expression is upregulated in the MPTP-treated animal model of PD [66] and that microglial TLR4 is directly activated by α-synuclein treatment [10]. Taken together, these results suggest that the modulation of microglial TLR4 by controlling pKr-2 production may provide us with important clues regarding the mechanisms responsible for inflammation-associated neurodegeneration in PD and open innovative therapeutic perspectives for the treatment of PD. For instance, treatment with minocycline has neuroprotective effects in preclinical studies of neurodegenerative diseases [2,30,67,68,69,70,71]. In particular, treatment with minocycline protects DA neurons against pKr-2-induced neurotoxicity through the inhibition of inflammatory responses in the brains of adult mice [2]. Moreover, its treatment reduces the expression of proinflammatory cytokines and iNOS, which is significantly increased by activated microglia following pKr-2 upregulation. These results suggest that the development of efficient anti-inflammatory drugs against pKr-2 may be useful for protecting DA neurons in the SN of lesioned adult brain.
Conclusion
- Go to
- Abstract
- INTRODUCTION
- Parkinson's disease and microglial activation
- Microglia and neuroinflammation
- Control of neuroin flammation by micro glial TLR4
- Production of pKr-2 and its general roles
in vivo - pKr-2 as an endogenous pathogen in the nigrostriatal DA system
- Control of pKr-2-induced neurotoxicity
- Conclusion
- Figure
- Reference
The discovery of endogenous biomolecules that initiate and stimulate microglial activation and result in neurodegeneration
Figures
- Go to
- Abstract
- INTRODUCTION
- Parkinson's disease and microglial activation
- Microglia and neuroinflammation
- Control of neuroin flammation by micro glial TLR4
- Production of pKr-2 and its general roles
in vivo - pKr-2 as an endogenous pathogen in the nigrostriatal DA system
- Control of pKr-2-induced neurotoxicity
- Conclusion
- Figure
- Reference
{kind=link}

{kind=link}
{kind=link}
References
- Go to
- Abstract
- INTRODUCTION
- Parkinson's disease and microglial activation
- Microglia and neuroinflammation
- Control of neuroin flammation by micro glial TLR4
- Production of pKr-2 and its general roles
in vivo - pKr-2 as an endogenous pathogen in the nigrostriatal DA system
- Control of pKr-2-induced neurotoxicity
- Conclusion
- Figure
- Reference
- Kim SR, Kareva T, Yarygina O, Kholodilov N, Burke RE. AAV transduction of dopamine neurons with constitutively active Rheb protects from neurodegeneration and mediates axon regrowth. Mol Ther 2012;20:275-286.

- Nam JH, Leem E, Jeon MT, Jeong KH, Park JW, Jung UJ, Kholodilov N, Burke RE, Jin BK, Kim SR. Induction of GDNF and BDNF by hRheb(S16H) transduction of SNpc neurons: neuroprotective mechanisms of hRheb(S16H) in a model of Parkinson's disease. Mol Neurobiol 2015;51:487-499.
- Shin WH, Jeon MT, Leem E, Won SY, Jeong KH, Park SJ, McLean C, Lee SJ, Jin BK, Jung UJ, Kim SR. Induction of microglial toll-like receptor 4 by prothrombin kringle-2: a potential pathogenic mechanism in Parkinson's disease. Sci Rep 2015;5:14764.
- Hirsch EC, Hunot S. Neuroinflammation in Parkinson's disease: a target for neuroprotection?. Lancet Neurol 2009;8:382-397.
- Kim SR, Chung ES, Bok E, Baik HH, Chung YC, Won SY, Joe E, Kim TH, Kim SS, Jin MY, Choi SH, Jin BK. Prothrombin kringle-2 induces death of mesencephalic dopaminergic neurons
in vivo andin vitro via microglial activation. J Neurosci Res 2010;88:1537-1548.
- Akira S, Uematsu S, Takeuchi O. Pathogen recognition and innate immunity. Cell 2006;124:783-801.
- Kagan JC, Medzhitov R. Phosphoinositide-mediated adaptor recruitment controls Toll-like receptor signaling. Cell 2006;125:943-955.
- Jin JJ, Kim HD, Maxwell JA, Li L, Fukuchi K. Toll-like receptor 4-dependent upregulation of cytokines in a transgenic mouse model of Alzheimer's disease. J Neuroinflammation 2008;5:23.
- Song M, Jin J, Lim JE, Kou J, Pattanayak A, Rehman JA, Kim HD, Tahara K, Lalonde R, Fukuchi K. TLR4 mutation reduces microglial activation, increases Aβ deposits and exacerbates cognitive deficits in a mouse model of Alzheimer's disease. J Neuroinflammation 2011;8:92.
- Fellner L, Irschick R, Schanda K, Reindl M, Klimaschewski L, Poewe W, Wenning GK, Stefanova N. Toll-like receptor 4 is required for alpha-synuclein dependent activation of microglia and astroglia. Glia 2013;61:349-360.
- Stefanova N, Reindl M, Neumann M, Kahle PJ, Poewe W, Wenning GK. Microglial activation mediates neurodegeneration related to oligodendroglial alpha-synucleinopathy: implications for multiple system atrophy. Mov Disord 2007;22:2196-2203.
- Dauer W, Przedborski S. Parkinson's disease: mechanisms and models. Neuron 2003;39:889-909.
- Kalia LV, Lang AE. Parkinson's disease. Lancet 2015;386:896-912.
- Katzenschlager R, Lees AJ. Treatment of Parkinson's disease: levodopa as the first choice. J Neurol 2002;249:II19-II24.
- Thanvi BR, Lo TC. Long term motor complications of levodopa: clinical features, mechanisms, and management strategies. Postgrad Med J 2004;80:452-458.
- Winklhofer KF, Haass C. Mitochondrial dysfunction in Parkinson's disease. Biochim Biophys Acta 2010;1802:29-44.
- Exner N, Lutz AK, Haass C, Winklhofer KF. Mitochondrial dysfunction in Parkinson's disease: molecular mechanisms and pathophysiological consequences. EMBO J 2012;31:3038-3062.
- Cui K, Luo X, Xu K, Ven Murthy MR. Role of oxidative stress in neurodegeneration: recent developments in assay methods for oxidative stress and nutraceutical antioxidants. Prog Neuropsychopharmacol Biol Psychiatry 2004;28:771-799.
- Wang Q, Liu Y, Zhou J. Neuroinflammation in Parkinson's disease and its potential as therapeutic target. Transl Neurodegener 2015;4:19.
- Imamura K, Hishikawa N, Sawada M, Nagatsu T, Yoshida M, Hashizume Y. Distribution of major histocompatibility complex class II-positive microglia and cytokine profile of Parkinson's disease brains. Acta Neuropathol 2003;106:518-526.
- Ouchi Y, Yoshikawa E, Sekine Y, Futatsubashi M, Kanno T, Ogusu T, Torizuka T. Microglial activation and dopamine terminal loss in early Parkinson's disease. Ann Neurol 2005;57:168-175.
- Hanisch UK. Microglia as a source and target of cytokines. Glia 2002;40:140-155.
- Gomes-Leal W. Microglial physiopathology: how to explain the dual role of microglia after acute neural disorders?. Brain Behav 2012;2:345-356.
- Frank-Cannon TC, Alto LT, McAlpine FE, Tansey MG. Does neuroinflammation fan the flame in neurodegenerative diseases?. Mol Neurodegener 2009;4:47.
- Kamata H, Hirata H. Redox regulation of cellular signalling. Cell Signal 1999;11:1-14.
- Haslund-Vinding J, McBean G, Jaquet V, Vilhardt F. NADPH oxidases in microglia oxidant production: activating receptors, pharmacology, and association with disease. Br J Pharmacol 2016
- Miller RL, James-Kracke M, Sun GY, Sun AY. Oxidative and inflammatory pathways in Parkinson's disease. Neurochem Res 2009;34:55-65.
- Wu DC, Teismann P, Tieu K, Vila M, Jackson-Lewis V, Ischiropoulos H, Przedborski S. NADPH oxidase mediates oxidative stress in the 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine model of Parkinson's disease. Proc Natl Acad Sci U S A 2003;100:6145-6150.
- Bové J, Perier C. Neurotoxin-based models of Parkinson's disease. Neuroscience 2012;211:51-76.
- Wu DC, Jackson-Lewis V, Vila M, Tieu K, Teismann P, Vadseth C, Choi DK, Ischiropoulos H, Przedborski S. Blockade of microglial activation is neuroprotective in the 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine mouse model of Parkinson disease. J Neurosci 2002;22:1763-1771.
- Członkowska A, Kohutnicka M, Kurkowska-Jastrzebska I, Członkowski A. Microglial reaction in MPTP (1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine) induced Parkinson's disease mice model. Neurodegeneration 1996;5:137-143.
- Cicchetti F, Brownell AL, Williams K, Chen YI, Livni E, Isacson O. Neuroinflammation of the nigrostriatal pathway during progressive 6-OHDA dopamine degeneration in rats monitored by immunohistochemistry and PET imaging. Eur J Neurosci 2002;15:991-998.
- Mosley RL, Benner EJ, Kadiu I, Thomas M, Boska MD, Hasan K, Laurie C, Gendelman HE. Neuroinflammation, oxidative stress and the pathogenesis of Parkinson's disease. Clin Neurosci Res 2006;6:261-281.
- Mogi M, Harada M, Riederer P, Narabayashi H, Fujita K, Nagatsu T. Tumor necrosis factor-alpha (TNF-α) increases both in the brain and in the cerebrospinal fluid from Parkinsonian patients. Neurosci Lett 1994;165:208-210.
- Kim HD, Jeong KH, Jung UJ, Kim SR. Myricitrin ameliorates 6-hydroxydopamine-induced dopaminergic neuronal loss in the substantia nigra of mouse brain. J Med Food 2016;19:374-382.
- Rodriguez-Pallares J, Parga JA, Muñoz A, Rey P, Guerra MJ, Labandeira-Garcia JL. Me chanism of 6-hydroxydopamine neurotoxicity: the role of NADPH oxidase and microglial activation in 6-hydroxydopamine-induced degeneration of dopaminergic neurons. J Neurochem 2007;103:145-156.
- Hernandes MS, Santos GD, Café-Mendes CC, Lima LS, Scavone C, Munhoz CD, Britto LR. Microglial cells are involved in the susceptibility of NADPH oxidase knockout mice to 6-hydroxy-dopamine-induced neurodegeneration. PLoS One 2013;8:e75532.
- Gao HM, Hong JS, Zhang W, Liu B. Distinct role for microglia in rotenone-induced degeneration of dopaminergic neurons. J Neurosci 2002;22:782-790.
- Gao HM, Liu B, Hong JS. Critical role for microglial NADPH oxidase in rotenone-induced degeneration of dopaminergic neurons. J Neurosci 2003;23:6181-6187.
- Molteni M, Gemma S, Rossetti C. The role of toll-like receptor 4 in infectious and noninfectious inflammation. Mediators Inflamm 2016;2016:6978936.
- Takeda K, Akira S. Toll-like receptors in innate immunity. Int Immunol 2005;17:1-14.
- Akira S, Takeda K. Toll-like receptor signalling. Nat Rev Immunol 2004;4:499-511.
- Olson JK, Miller SD. Microglia initiate central nervous system innate and adaptive immune responses through multiple TLRs. J Immunol 2004;173:3916-3924.
- Lehnardt S, Massillon L, Follett P, Jensen FE, Ratan R, Rosenberg PA, Volpe JJ, Vartanian T. Activation of innate immunity in the CNS triggers neurodegeneration through a Toll-like receptor 4-dependent pathway. Proc Natl Acad Sci U S A 2003;100:8514-8519.
- Ahmad A, Crupi R, Campolo M, Genovese T, Esposito E, Cuzzocrea S. Absence of TLR4 reduces neurovascular unit and secondary inflammatory process after traumatic brain injury in mice. PLoS One 2013;8:e57208.
- Caso JR, Pradillo JM, Hurtado O, Lorenzo P, Moro MA, Lizasoain I. Toll-like receptor 4 is involved in brain damage and inflammation after experimental stroke. Circulation 2007;115:1599-1608.
- Kerfoot SM, Long EM, Hickey MJ, Andonegui G, Lapointe BM, Zanardo RC, Bonder C, James WG, Robbins SM, Kubes P. TLR4 contributes to disease-inducing mechanisms resulting in central nervous system autoimmune disease. J Immunol 2004;173:7070-7077.
- Walter S, Letiembre M, Liu Y, Heine H, Penke B, Hao W, Bode B, Manietta N, Walter J, Schulz-Schuffer W, Fassbender K. Role of the toll-like receptor 4 in neuroinflammation in Alzheimer's disease. Cell Physiol Biochem 2007;20:947-956.
- Gambuzza ME, Sofo V, Salmeri FM, Soraci L, Marino S, Bramanti P. Toll-like receptors in Alzheimer's disease: a therapeutic perspective. CNS Neurol Disord Drug Targets 2014;13:1542-1558.
- Noelker C, Morel L, Lescot T, Osterloh A, Alvarez-Fischer D, Breloer M, Henze C, Depboylu C, Skrzydelski D, Michel PP, Dodel RC, Lu L, Hirsch EC, Hunot S, Hartmann A. Toll like receptor 4 mediates cell death in a mouse MPTP model of Parkinson disease. Sci Rep 2013;3:1393.
- Park HS, Jung HY, Park EY, Kim J, Lee WJ, Bae YS. Cutting edge: direct interaction of TLR4 with NAD(P)H oxidase 4 isozyme is essential for lipopolysaccharide-induced production of reactive oxygen species and activation of NF-kappa B. J Immunol 2004;173:3589-3593.
- Dihanich M, Kaser M, Reinhard E, Cunningham D, Monard D. Prothrombin mRNA is expressed by cells of the nervous system. Neuron 1991;6:575-581.
- Mann KG. Prothrombin. Methods Enzymol 1976;45:123-156.
- Taneda H, Andoh K, Nishioka J, Takeya H, Suzuki K. Blood coagulation factor Xa interacts with a linear sequence of the kringle 2 domain of prothrombin. J Biochem 1994;116:589-597.
- Shikamoto Y, Morita T. Expression of factor X in both the rat brain and cells of the central nervous system. FEBS Lett 1999;463:387-389.
- Lee TH, Rhim T, Kim SS. Prothrombin kringle-2 domain has a growth inhibitory activity against basic fibroblast growth factor-stimulated capillary endothelial cells. J Biol Chem 1998;273:28805-28812.
- Rhim TY, Park CS, Kim E, Kim SS. Human prothrombin fragment 1 and 2 inhibit bFGF-induced BCE cell growth. Biochem Biophys Res Commun 1998;252:513-516.
- Kim TH, Oh S, Kim SS. Recombinant human prothrombin kringle-2 induces bovine capillary endothelial cell cycle arrest at G0-G1 phase through inhibition of cyclin D1/CDK4 complex: modulation of reactive oxygen species generation and up-regulation of cyclin-dependent kinase inhibitors. Angiogenesis 2005;8:307-314.
- Dasgupta SK, Thiagarajan P. Inhibition of thrombin activity by prothrombin activation fragment 1.2. J Thromb Thrombolysis 2007;24:157-162.
- Kim TH, Ahn S, Kim J, Kim I, Yang MZ, Lee JE, Kim SS. Recombinant human prothrombin kringle-2 inhibits B16F10 melanoma metastasis through inhibition of neovascularization and reduction of matrix metalloproteinase expression. Clin Exp Metastasis 2006;23:391-399.
- Ishida Y, Nagai A, Kobayashi S, Kim SU. Upregulation of protease-activated receptor-1 in astrocytes in Parkinson disease: astrocyte-mediated neuroprotection through increased levels of glutathione peroxidase. J Neuropathol Exp Neurol 2006;65:66-77.
- Berzin TM, Zipser BD, Rafii MS, Kuo-Leblanc V, Yancopoulos GD, Glass DJ, Fallon JR, Stopa EG. Agrin and microvascular damage in Alzheimer's disease. Neurobiol Aging 2000;21:349-355.
- Sokolova E, Reiser G. Prothrombin/thrombin and the thrombin receptors PAR-1 and PAR-4 in the brain: localization, expression and participation in neurodegenerative diseases. Thromb Haemost 2008;100:576-581.
- Won SY, Choi SH, Jin BK. Prothrombin kringle-2-induced oxidative stress contributes to the death of cortical neurons
in vivo andin vitro : role of microglial NADPH oxidase. J Neuroimmunol 2009;214:83-92.
- Ryu J, Min KJ, Rhim TY, Kim TH, Pyo H, Jin B, Kim SU, Jou I, Kim SS, Joe EH. Prothrombin kringle-2 activates cultured rat brain microglia. J Immunol 2002;168:5805-5810.
- Ros-Bernal F, Hunot S, Herrero MT, Parnadeau S, Corvol JC, Lu L, Alvarez-Fischer D, Carrillo-de Sauvage MA, Saurini F, Coussieu C, Kinugawa K, Prigent A, Höglinger G, Hamon M, Tronche F, Hirsch EC, Vyas S. Microglial glucocorticoid receptors play a pivotal role in regulating dopaminergic neurodegeneration in Parkinsonism. Proc Natl Acad Sci U S A 2011;108:6632-6637.
- Choi SH, Lee DY, Chung ES, Hong YB, Kim SU, Jin BK. Inhibition of thrombin-induced microglial activation and NADPH oxidase by minocycline protects dopaminergic neurons in the substantia nigra
in vivo . J Neurochem 2005;95:1755-1765.
- Noble W, Garwood C, Stephenson J, Kinsey AM, Hanger DP, Anderton BH. Minocycline reduces the development of abnormal tau species in models of Alzheimer's disease. FASEB J 2009;23:739-750.
- Chen M, Ona VO, Li M, Ferrante RJ, Fink KB, Zhu S, Bian J, Guo L, Farrell LA, Hersch SM, Hobbs W, Vonsattel JP, Cha JH, Friedlander RM. Minocycline inhibits caspase-1 and caspase-3 expression and delays mortality in a transgenic mouse model of Huntington disease. Nat Med 2000;6:797-801.
- Arvin KL, Han BH, Du Y, Lin SZ, Paul SM, Holtzman DM. Minocycline markedly protects the neonatal brain against hypoxic-ischemic injury. Ann Neurol 2002;52:54-61.
- Sanchez Mejia RO, Ona VO, Li M, Friedlander RM. Minocycline reduces traumatic brain injury-mediated caspase-1 activation, tissue damage, and neurological dysfunction. Neurosurgery 2001;48:1393-1399.