Articles

Article Tools
 View Full Text View Full Text |
 Abstract Abstract |
 Article as PDF Article as PDF |
 Print this Article Print this Article |
 Pubmed Pubmed |
 PMC PMC |
 PubReader PubReader |
 Export to Citation Export to Citation |
 Email Alerts Email Alerts |
 Open Access Open Access |
Share this article on :



Stats or Metrics
Article
Original Article
Exp Neurobiol 2013; 22(1): 51-57
Published online March 30, 2013
https://doi.org/10.5607/en.2013.22.1.51
© The Korean Society for Brain and Neural Sciences
TRPV1 Activation in Primary Cortical Neurons Induces Calcium-Dependent Programmed Cell Death
Juhyun Song1, Jun Hong Lee1, Sung Ho Lee1, Kyung Ah Park1, Won Taek Lee1* and Jong Eun Lee1,2*
1Department of Anatomy, 2BK21 Project for Medical Sciences, Yonsei University College of Medicine, Seoul 120-752, Korea
Correspondence to: *To whom correspondence should be addressed.
Jong Eun Lee
TEL: 82-2-2228-1646, 1659, FAX: 82-2-365-0700
e-mail: jelee@yuhs.ac
Won Taek Lee
TEL: 82-2-2228-1644, 1659, FAX: 82-2-365-0700
e-mail: INSKULL@yuhs.ac
Transient receptor potential cation channel, subfamily V, member 1 (TRPV1, also known as vanilloid receptor 1) is a receptor that detects capsaicin, a pungent component of chili peppers, and noxious heat. Although its function in the primary nociceptor as a pain receptor is well established, whether TRPV1 is expressed in the brain is still under debate. In this study, the responses of primary cortical neurons were investigated. Here, we report that 1) capsaicin induces caspase-3-dependent programmed cell death, which coincides with increased production of nitric oxide and peroxynitrite ; that 2) the prolonged capsaicin treatment induces a steady increase in the degree of capase-3 activation, which is prevented by the removal of capsaicin; 3) and that blocking calcium entry and calcium-mediated signaling prevents capsaicin-induced cell death. These results indicate that cortical neurons express TRPV1 whose prolonged activation causes cell death.
Keywords: capsaicin, primary neuron, apoptosis, Ca2+, NOS, caspase-3
INTRODUCTION
Capsaicin (8-methyl-N-vanillyl-6-nonenmide) from red peppers elicits painful sensation by directly activating TRPV1 expressed in the unmyelinated C fibers or the small myelinated Aδ fiber of primary sensory neurons [1]. After cloning the TRPV1 as the capsaicin receptor, many highly related members of TRP channel families have been identified [2-4]. To date TRPV1 is the only channel that can be activated by capsaicin. TRPV1 is mainly expressed by nociceptors, the specialized subset of primary sensory neurons in the dorsal root and trigeminal ganglia dedicated to the detection of noxious stimuli, and is activated by vanilloid ligands such as capsaicin, noxious heat or acid [5]. Accumulating evidence suggests that TRPV1 is also expressed across the central nervous system such as in the forebrain, the midbrain tegmentum, the hindbrain and the hypothalamus, and prolonged capsaicin treatment to animals induces degeneration in these brain regions [6]. Although the expression of TRPV1 in the central nervous system (CNS) has been suggested by many studies [7], it is still under debate to which degree the expression data can be functionally relevant as many of these "TRPV1-positive" do not respond to capsaicin in calcium imaging assay [8]. Indeed, It is still unclear whether TRPV1 signaling is a prevalent mode to detect noxious stimuli in the brain, as capsaicin produces varying effects depending on the developmental stage and the location [9]. Although TRPV1 is a non-selective cation channel, calcium in the most important ion that mediates capsaicin-mediated cell death [10]. Sustained elevation in intracellular calcium concentraion activates various secondary mechanisms that induces the increased production of reactive oxygen and nitrogen species and ultimately the programmed cell death [11, 12]. In this study, we show that TRPV1 is expressed in primary cortical neurons and that capsaicin induces capasae 3-dependent programmed cell death, which can be prevented by inhibiting calcium-mediated signaling.
MATERIALS AND METHODS
The ICR mice were purchased from Samtako Co. (Kyunggi, Korea). Sodium bicarbonate, HEPES, glutamine and MEM medium (#330-1430) were obtained from Gibco BRL (Grand Island, NY), and fetal bovine (FBS) and horse serum supplied by Hyclone Co. (Logan, UT). Capsaicin, agmatine, NADPH, nitroblue tetrazolium, CoCl2 and Griess reagent were obtained from Sigma Chemical Co. (St Louis, MO). The anti-capsaicin receptor sera and the anti-nNOS sera were purchased from Oncogene Research Products (Cambridge, MA). All the plastic ware was bought from Nunc Co. (Naperville, IL). Other chemicals were of research grade.
Embryonic pup brains were extracted from rat (E17-19) and placed in a petri dish containing HBSS. The tissue were transfered to 50 ml sterile tube and then were allowed to settle to bottom of tube and aspirate off excess HBSS (Hank's buffered salt solution) using 5 ml pipet. Next, we add 30 ml trypsin (Sigma) to tube containing tissue, and place in a 37℃ water bath for 17 minutes. In that containing tube, add 1 ml FBS and mix by inverting tube. With a 5 ml pipet, aspirate as much of the FBS off of tissue as possible taking care not to aspirate any tissue. And we wash 1-2 times with plain HBSS. To each tube of washed tissue, add 5 ml HBSS and pipet up and down to break up tissue. And then divide the above 5 ml tissue suspension between two 15 ml tubes, and triturate suspension with sterile 9 Pasteur pipet. Filter cell suspension from each tube through a 40 micron nylon cell strainer (Fisher). And spin filtered cell suspension. Cell lysis and subsequent release of genomic DNA may lead to the formation of tissue clots. Resuspend pellet in full plating media (Neurobasal/B-27/L-glutamine/antibiotic). And also resuspend in about 2 ml per embryonic brain. Next, take 20 µl of resuspended culture and add 1 µl trypan blue. Count viable (round and bright) cells, and plate on poly-D-lysine treated plasticware. And plate cells at low density for imaging (3×105 cells/35 dish with coverslip; 2×105 cells/well of 6-well plate), or plate at a high density for biochemistry (3×106 cells/well of 6-well plate) as soon as possible. Cultures maintained for at least one week to allow for differentiation. Cultures were maintained in a humidified atmosphere of 95% air and 5% CO2 at 37℃. Culture media were replaced totally every 3 days. NSCs of 2 week culture were used for the experiment.
Neuronal cell death was investigated in a neuron culture treated with capsaicin (final concentration: 10, 100 µM). The culture media was changed with a balanced salt solution 5.5 (160 mM NaCl, 5.36 mM KCl, 0.81 mM MgSO4, 1 mM NaH2PO4, 10 mM HEPES, 1.8 mM CaCl2, 14.7 mM NaHCO3, 5.5 mM glucose, pH7.4) prior to capsaicin treatment. In order to inhibit the NOS activity, agmatine was added in the culture at 100 mM, and then incubated for 12 h prior to capsaicin treatment. In terms of the caspase inhibitor, the neurons were pre-incubated 1 h before the capsaicin was added. In the experiments associated with calcium, the calcium free condition was prepared by excluding calcium in the BSS5.5 media, and the calcium channel blocker, Nifedipine, was added to the BSS5.5 media at 1 µM. EGTA, cation-chelating agent, was added in the culture at 50 µM.
Based on the form of the nucleus fragmentation and the loss of the plasma membrane, apoptotic cells can be discriminated from non-apoptotic cells using a propidium iodide and Hoechst 33258 staining solution (Sigma Co., St Louis, MA). Highly charged propidium iodide cannot enter intact cells whereas Hoechst can pass through either an intact or dead cell membrane. Consequently, the cells were stained with the Hoechst-propidium iodide to measure the number of apoptotic and necrotic cells and to calculate each cell. The Hoechst stain solution was added into the growth media at 2-5 µg/ml, and incubated for 30 min at 37℃. Subsequently, the propidium iodide solution was added into the media at the same concentration as the Hoechst stain. The extent of cell damage was observed by fluorescent microscopy (Olympus microscope, Japan).
The release of LDH is a widely used index of cellular injury. The amount of total LDH released from 100% cell deathnamed "full kill" was determined at the end of the experimentby freezing the NSCs at -70℃ and rapid thawing. The extentof cell death was expressed as percentage of full kill. LDH% was calculated against the percentage of full kill.
After incubating the primary cultured neurons in a 24-well plate, the culture media was replaced by BSS5.5 media. The neuronal culture was treated with capsaicin, which was then harvested at the appropriate times. The caspase-3 activity was measuredas described in the procedure of Blom et al. [13]. The neurons were lysed in a 100 µl cell lysis buffer by freezing at -20℃ followed by rapid thawing. The lysate was centrifuged at 15,000 × g for 20 min, and 20 µl of the supernatant was used for incubation with 2 µl DEVD-pNA (Calbiochem, La Jolla, CA, USA). The reaction mixture was incubated at 37℃ for 3 h, and then measured at 405 nm by a plate reader. The specific caspase-3 activity was calculated from the relative absorbance of the sample to a control using the standard pNA curve. The protein concentration was determined by a BCA protein assay solution (Pierce, Rockford, IL, USA).
In order to determine the level of nitrite production in the neuron cultures, each culture treated with capsaicin and agmatine (each 100 µl) was transferred to 96-well plates. The Griess reagents (100 ml) were added to the plate and incubated for 15 min at room temperature. Subsequently, the absorbance was measured at 540 nm. Standards were prepared with nitrite solution instead of the sample [14]. The nitrite concentration in the sample was calculated by comparing the absorbance of the sample with a nitrite-standard curve.
The capsaicin receptor and nNOS expression distribution in the CNS neurons were investigated by immunohistochemistry using the avidin-biotin-peroxidase complex (ABC) method using mouse polyclonal anti-capsaicin receptor sera and anti-nNOS sera. The cultured primary pure neurons were reacted with polyclonal anti-capsaicin receptor sera (Dilution: 2 µg/ml) as the first antibody. The biotin labeled immunoglobulin and avidin-biotin-peroxidase complex, which specifically bound to the 2nd antibody were used as a 2nd and 3rd reagent, respectively. Finally, a mixed solution of 0.05% 3,3'-diaminobenzidine-tetrahydrochloride and 0.01% H2O2 was used as the developing solution, and was observed by optical microscopy. In terms of nNOS expression, the above procedure was used except for the use of polyclonal anti-nNOS sera.
Lactate dehydrogenase (LDH) is an enzyme that catalyzes the interconversion of pyruvate and lactate with concomitant interconversion of NADH. LDH is expressed in almost all cells and its release is a widely used indicator of tissue damage and cell death. Treating capsaicin for 24 hours resulted in high increase in LDH release from cultured cortical and hippocampal neurons indicating cell death (Fig. 1A). Concomitantly, capsaicin-treated cells showed an increased the level of active caspase-3 (Fig. 1B) and apoptotic morphology as evidenced by propidium iodine-Hoeschst assay (Fig. 1C-F), suggesting that prolonged capsaicin treatment induces caspase-3-dependent apoptosis in these cells.
Caspase-3 is the executioner of apoptosis, whose signaling requires Ca2+. Having established that capsaicin treatment induces apoptosis in primary cortical and hippocampal neurons, we investigated mechanisms underlying capsaicin-induced cell death using cortical neurons as a model. First we determined the time course of capsaicin-induced caspase-3 activation. The level of active casepase-3 increased sharply in 6 hours of capsaicin treatment and remained high for the duration of the study (24 hours) (Fig. 2). Withdrawing capsaicin after 12 h treatment decreased the overall activity of caspase-3, which returned to the baseline level in additional 12 hours (Fig. 2). These results indicate that sustained capsaicin treatment exerts incremental effects on cell death and suggest that inhibiting capsaicin- and TRPV1-mediated signaling can reverse capsaicin-mediated toxicity.
We previously showed that capsaicin induces apoptosis of cortical neurons that express TRPV1 [9]. Although TRPV1 is a non-selective cation channel and thus capsaicin treatment induces the flux of several ions such as Na+ and Ca2+, Ca2+ is a key mediator of apoptosis. Therefore, we investigated whether blocking various stages of calcium signaling affects capsaicin-mediated apoptosis. We pre-treated the cortical neurons with Nifedipine, a blocker of voltage-gated L-type calcium channel, calcium-free medium, or medium containing 50 µM EGTA- a calcium chelator, before capsaicin treatment. The cells pretreated with Nifedipine and cultured in calcium free media showed a reduction in apoptotic cell death (Fig. 3E). In terms of EGTA treatment, the neurons were protected from cell death using a LDH assay whereas apoptotic patterns of the nuclei were observed in most cells by Hoechst-PI staining. These results suggested that apoptosis is induced in the cells pretreated with EGTA. However, membrane damage is insufficient to release adequate amounts of LDH protein into the culture media. Whereas, the neurons pretreated with Nifedipine showed no apoptosis, which is similar to those of the LDH assay (Fig. 3A).
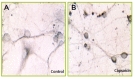
Nitric oxide (NO) is a key mediator that links increased calcium signaling to apoptosis. We reasoned that capsaicin treatment will activate an calcium- and NO-dependent apoptosis in cortical neurons. We measured the expression and activity of nitrix oxide synthase (NOS) by neuronal NOS (nNOS) immunohistochemistry (Fig. 4). Capsaicin treatment increased both nNOS expression, indicating that capsaicin activates a signaling cascade that results in the increased production of NO (Fig. 4).
Peroxynitrite (ONOO-) is an oxidant and nitrating agent, which can damage DNA and proteins, and is a useful indicator of oxidative stress. We measured peroxynitrite (ONOO-) in cortical neurons and found that capsaicin treatment increases it. This result is consistent with the previous results of this manuscript indicating that prolonged capsaicin treatment induces calcium channel mediated apoptosis (Fig. 5).
Our results indicate that capsaicin can induce cell death in primary cultured neurons by apoptosis consistent with previous reports by Sugimoto et al. [15, 16]. In the nervous system, Caspase-3 and caspase-9 are activated in neurons following peripheral nerve injury, ischemia [17-19]. Whereas caspase can be activated by extrinsic and intrinsic signals, whether capsaicin stimulation directly activates caspases has not been demonstrated. Our data showing that capsaicin induces LDH release and apoptotic nuclear morphology support this possibility. Ca2+ is a key mediator of apoptosis [3, 20]. Capsaicin activates Ca2+ signaling by promoting Ca2+ influx through TRPV1 channels [21-24], which leads to p38- and oxidative stress-induced cell death [25]. Capsaicin stimulation preferentially activates other Ca2+-dependent pathways such as caspase signaling [26, 27], as TRPV1 has higher conductance of Ca2+ compared to those of Mg2+, Na+ or other cations [28]. In this study, we investigated how capsaicin-induced apoptosis in different primary neurons is affected by Ca2+ and found that preventing a rise in intracellular Ca2+ attenuates capsaicin-induced cell death. Mediators of apoptosis downstream of Ca2+ include caspases and nitric oxide (NO). NO, whose production is regulated by nitrix oxide synthase (NOS), can cause multiple effects such as energy depletion-induced necrosis and apoptosis [29]. In particular, a high concentration of NO inhibits mitochondrial cytochrome oxidase in neurons and inhibits mitochondrial function [30, 31]. Neuronal NOS (nNOS) is a neuron-specific subtype of which produces NO using L-arginine and oxygen as a substrate [32, 33]. NO plays important roles as a signaling molecule; a vasodilator in the vascular system, a defensive molecule in phagocytes, and a cell signaling molecule that activates guanylyl cyclase and increases the amount of cGMP as an intercellular messenger. NO and NO metabolites known as reactive nitrogen species (RNS) ultimately induce the oxidation, nitration (addition of NO2), nitrosation (addition of NO+), and nirosylation (addition of NO) of most biomolecules [34]. This study demonstrates that capsaicin induced the activation of NO through the augmentation of nNOS expression and NOS activity. Several researches provided that NO exerts cytotoxicity when it is transformed to peroxynitrate [28, 35, 36]. In this study, we assessed the amount of peroxynitrate by measuring the relative amount of protein nitration and showed that capsaicin stimulation induces an increase in peroxynitrate in cortical neurons. Together, our results indicate that capsaicin, acting directly on TRPV1 expressed in primary cortical neurons, induces Ca2+-dependent apoptosis by mechanisms that involve casapse 3 activation, NO synthesis, and an increased production of peroxynitrate. As TRPV1 is activated by high temperature (42C), our data not only suggest that a sustained hyperthermia may induce irreversible damage to the central nervous system by increase neuronal apoptosis, but also that promptly lowering an elevated body temperature may be the efficient way to prevent further damage. Further research on the relationship between NO and caspase pathways will reveal more sights into TRPV1-induced cell death in the central nervous system.




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
- Hayes AG, Tyers MB. Effects of capsaicin on nociceptive heat, pressure and chemical thresholds and on substance P levels in the rat. Brain Res 1980;189:561-564.

- Caterina MJ, Rosen TA, Tominaga M, Brake AJ, Julius D. A capsaicin-receptor homologue with a high threshold for noxious heat. Nature 1999;398:436-441.
- Caterina MJ, Schumacher MA, Tominaga M, Rosen TA, Levine JD, Julius D. The capsaicin receptor: a heat-activated ion channel in the pain pathway. Nature 1997;389:816-824.
- Suzuki M, Sato J, Kutsuwada K, Ooki G, Imai M. Cloning of a stretch-inhibitable nonselective cation channel. J Biol Chem 1999;274:6330-6335.
- Liu L, Simon SA. Capsaicin, acid and heat-evoked currents in rat trigeminal ganglion neurons: relationship to functional VR1 receptors. Physiol Behav 2000;69:363-378.
- Ritter S, Dinh TT. Capsaicin-induced neuronal degeneration: silver impregnation of cell bodies, axons, and terminals in the central nervous system of the adult rat. J Comp Neurol 1988;271:79-90.
- Mezey E, Tóth ZE, Cortright DN, Arzubi MK, Krause JE, Elde R, Guo A, Blumberg PM, Szallasi A. Distribution of mRNA for vanilloid receptor subtype 1 (VR1), and VR1-like immunoreactivity, in the central nervous system of the rat and human. Proc Natl Acad Sci U S A 2000;97:3655-3660.
- Cavanaugh DJ, Chesler AT, Jackson AC, Sigal YM, Yamanaka H, Grant R, O'Donnell D, Nicoll RA, Shah NM, Julius D, Basbaum AI. Trpv1 reporter mice reveal highly restricted brain distribution and functional expression in arteriolar smooth muscle cells. J Neurosci 2011;31:5067-5077.
- Lee WT, Lee JH, Lee SH, Park KA, Lee JE. Differential effect of capsaicin on the cell death in primary cultured neurons. Korean J Anat 2002;35:161-171.
- Holzer P. Capsaicin: cellular targets, mechanisms of action, and selectivity for thin sensory neurons. Pharmacol Rev 1991;43:143-201.
- Rego AC, Oliveira CR. Mitochondrial dysfunction and reactive oxygen species in excitotoxicity and apoptosis: implications for the pathogenesis of neurodegenerative diseases. Neurochem Res 2003;28:1563-1574.
- Forder JP, Tymianski M. Postsynaptic mechanisms of excitotoxicity: involvement of postsynaptic density proteins, radicals, and oxidant molecules. Neuroscience 2009;158:293-300.
- Blom WM, de Bont HJ, Meijerman I, Mulder GJ, Nagelkerke JF. Prevention of cycloheximide-induced apoptosis in hepatocytes by adenosine and by caspase inhibitors. Biochem Pharmacol 1999;58:1891-1898.
- Schmidt HH, Warner TD, Nakane M, Förstermann U, Murad F. Regulation and subcellular location of nitrogen oxide synthases in RAW264.7 macrophages. Mol Pharmacol 1992;41:615-624.
- Sugimoto T, Takeyama A, Xiao C, Takano-Yamamoto T, Ichikawa H. Electron microscopic demonstration of nick end-labeled DNA fragments during capsaicin-induced apoptosis of trigeminal primary neurons in neonatal rats. Brain Res 1999;818:147-152.
- Sugimoto T, Xiao C, Ichikawa H. Neonatal primary neuronal death induced by capsaicin and axotomy involves an apoptotic mechanism. Brain Res 1998;807:147-154.
- Cao G, Luo Y, Nagayama T, Pei W, Stetler RA, Graham SH, Chen J. Cloning and characterization of rat caspase-9: implications for a role in mediating caspase-3 activation and hippocampal cell death after transient cerebral ischemia. J Cereb Blood Flow Metab 2002;22:534-546.
- Sugimoto T, Jin H, Fijita M, Fukunaga T, Nagaoka N, Yamaai T, Ichikawa H. Induction of activated caspase-3-immunoreactivity and apoptosis in the trigeminal ganglion neurons by neonatal peripheral nerve injury. Brain Res 2004;1017:238-243.
- Kuan CY, Roth KA, Flavell RA, Rakic P. Mechanisms of programmed cell death in the developing brain. Trends Neurosci 2000;23:291-297.
- Nicotera P, Orrenius S. The role of calcium in apoptosis. Cell Calcium 1998;23:173-180.
- Caterina MJ, Schumacher MA, Tominaga M, Rosen TA, Levine JD, Julius D. The capsaicin receptor: a heat-activated ion channel in the pain pathway. Nature 1997;389:816-824.
- Premkumar LS, Ahern GP. Induction of vanilloid receptor channel activity by protein kinase C. Nature 2000;408:985-990.
- Sikand P, Premkumar LS. Potentiation of glutamatergic synaptic transmission by protein kinase C-mediated sensitization of TRPV1 at the first sensory synapse. J Physiol 2007;581:631-647.
- Tominaga M, Caterina MJ. Thermosensation and pain. J Neurobiol 2004;61:3-12.
- Amantini C, Mosca M, Nabissi M, Lucciarini R, Caprodossi S, Arcella A, Giangaspero F, Santoni G. Capsaicin-induced apoptosis of glioma cells is mediated by TRPV1 vanilloid receptor and requires p38 MAPK activation. J Neurochem 2007;102:977-990.
- Dedov VN, Roufogalis BD. Mitochondrial calcium accumulation following activation of vanilloid (VR1) receptors by capsaicin in dorsal root ganglion neurons. Neuroscience 2000;95:183-188.
- Leonelli M, Martins DO, Britto LR. TRPV1 receptors modulate retinal development. Int J Dev Neurosci 2011;29:405-413.
- Eliasson MJ, Huang Z, Ferrante RJ, Sasamata M, Molliver ME, Snyder SH, Moskowitz MA. Neuronal nitric oxide synthase activation and peroxynitrite formation in ischemic stroke linked to neural damage. J Neurosci 1999;19:5910-5918.
- Brown GC. Nitric oxide and neuronal death. Nitric Oxide 2010;23:153-165.
- Brown GC, Cooper CE. Nanomolar concentrations of nitric oxide reversibly inhibit synaptosomal respiration by competing with oxygen at cytochrome oxidase. FEBS Lett 1994;356:295-298.
- Bal-Price A, Matthias A, Brown GC. Stimulation of the NADPH oxidase in activated rat microglia removes nitric oxide but induces peroxynitrite production. J Neurochem 2002;80:73-80.
- Griffith OW, Stuehr DJ. Nitric oxide synthases: properties and catalytic mechanism. Annu Rev Physiol 1995;57:707-736.
- Klatt P, Heinzel B, John M, Kastner M, Böhme E, Mayer B. Ca2+/calmodulin-dependent cytochrome c reductase activity of brain nitric oxide synthase. J Biol Chem 1992;267:11374-11378.
- Patel RP, McAndrew J, Sellak H, White CR, Jo H, Freeman BA, Darley-Usmar VM. Biological aspects of reactive nitrogen species. Biochim Biophys Acta 1999;1411:385-400.
- Brunelli L, Crow JP, Beckman JS. The comparative toxicity of nitric oxide and peroxynitrite to Escherichia coli. Arch Biochem Biophys 1995;316:327-334.
- Huie RE, Padmaja S. The reaction of no with superoxide. Free Radic Res Commun 1993;18:195-199.