Articles

Article Tools
 View Full Text View Full Text |
 Abstract Abstract |
 Article as PDF Article as PDF |
 Print this Article Print this Article |
 Pubmed Pubmed |
 PMC PMC |
 PubReader PubReader |
 Export to Citation Export to Citation |
 Email Alerts Email Alerts |
 Open Access Open Access |
Share this article on :



Stats or Metrics
Article
Original Article
Exp Neurobiol 2023; 32(5): 354-361
Published online October 31, 2023
https://doi.org/10.5607/en23021
© The Korean Society for Brain and Neural Sciences
Methylation-based Subclassifications of Embryonal Tumor with Multilayered Rosettes in Not Just Pediatric Brains
Eric Eunshik Kim1, Kwanghoon Lee1, Ji-Hoon Phi2, Min-Sung Kim2, Hyoung Jin Kang3,4, Hongseok Yun5 and Sung-Hye Park1,6*
1Department of Pathology, College of Medicine, Seoul National University, Seoul 03080, 2Department of Neurosurgery, College of Medicine, Seoul National University, Seoul 03080, 3Department of Pediatrics, College of Medicine, Seoul National University, Seoul 03080, 4Cancer Research Institute, Seoul National University Children's Hospital, Seoul 03080, 5Department of Genomic Medicine, College of Medicine, Seoul National University Hospital, Seoul 03080, 6Institute of Neuroscience, College of Medicine, Seoul National University, Seoul 03080, Korea
Correspondence to: *To whom correspondence should be addressed.
TEL: 82-2-2072-3090, FAX: 82-2-743-5530
e-mail: shparknp@snu.ac.kr
This is an Open Access article distributed under the terms of the Creative Commons Attribution Non-Commercial License (http://creativecommons.org/licenses/by-nc/4.0) which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
Abstract
The aim of this study is to investigate the genetic profiles and methylation-based classifications of Embryonal tumor with multilayered rosettes (ETMR), with a specific focus on differentiating between C19MC amplified and C19MC-not amplified groups, including cases with DICER1 mutations. To achieve this, next-generation sequencing using a targeted gene panel for brain tumors and methylation class studies using the Epic850K microarray were performed to identify tumor subclasses and their clinicopathological characteristics. The study cohort consisted of four patients, including 3 children (a 4-months/F, a 9-months/M, and a 2 y/F), and one adult (a 30 y/Male). All three tumors in the pediatric patients originated in the posterior fossa and exhibited TTYH1:C19MC fusion and C19MC amplification. The fourth case in the adult patient involved the cerebellopontine angle with biallelic DICER1 mutation. Histopathological examination revealed typical embryonal features characterized by multilayered rosettes and abundant neuropils in all cases, while the DICER1-mutant ETMR also displayed cartilage islands in addition to the classic ETMR pathology. All four tumors showed positive staining for LIN28A. The t-SNE clustering analysis demonstrated that the first three cases clustered with known subtypes of ETMR, specifically C19MC amplified, while the fourth case clustered separately to non-C19MC amplified subclass. During the follow-up period of 6~12 months, leptomeningeal dissemination of the tumor occurred in all patients. Considering the older age of onset in DICER1-mutant ETMR, genetic counseling should be recommended due to the association of DICER1 mutations with germline and second-hit somatic mutations in cancer.
Graphical Abstract
Keywords: Brain neoplasm, Cerebral primitive neuroectodermal tumor, DNA methylation, DICER1
INTRODUCTION
Embryonal tumor with multilayered rosettes (ETMR) is a rare tumor type found in young children's central nervous system (CNS). Despite its infrequency, the grim prognosis associated with ETMR has drawn attention from both neuropathologists and oncologists, emphasizing the need for improved diagnostic and therapeutic approaches for this condition. While there is an immunohistochemical test available, such as LIN28A, it is not exclusive to ETMR. ETMR has less than a decade of diagnostic experience, which contributes to its challenging diagnosis and subclassification [1]. This tumor comprises a conglomerate of three morphologically distinct tumor types; embryonal tumor with abundant neuropil and true rosettes ependymoblastoma (ETANTR), ependymoblastoma (EBL), and medulloepithelioma (MEPL) [2]. The underlying molecular biology of ETMR primarily involves the
MATERIALS AND METHODS
Clinicopathologic assessment
After Institutional Review Board (IRB) approval, a clinical database search was undertaken to identify all diagnosed ETMR between 2016 and 2023 at Seoul National University Hospital (SNUH). Clinical and follow-up information obtained from institutional databases or electronic medical records included age, gender, radiologic assessments, types of surgery, medical management, and vital status at follow-up. At the time of diagnosis, LIN28A IHC was used as a screening tool when the morphology of the tumor suggested ETMR.
Molecular analyses with next-generation sequencing
The molecular genetics integrated diagnosis followed using a next-generation sequencing (NGS) panel called “FiRST Brain Tumor Panel (BTP)”, customized by SNUH as described in Lee et al [5]. The panel includes 14 essential genes and 210 selected genes for DNA samples, as well as 151 genes for RNA samples. Whole genome sequencing results were analyzed for chromosomal structural variations (SVs). Arriba was used to detect and visualize gene fusions from RNA sequencing data.
Methylation array analysis and t-SNE clustering with the Illumina 850K microarray
DNA methylation array analysis was performed for three cases of ETMR using an Infinium MethylationEPIC 850K BeadChip array platform (Illumina, USA). DNA methylation data analysis was performed using R software (R 4.2.0, https://www.r-project.org/). Our samples were analyzed using the methylation classifier of DKFZ with GSE90496, GSE122038 (2,979 samples from 92 subclasses) [3, 6]. A preprocessing procedure was conducted for raw methylation intensity signals using the R package “methylationArrayAnalysis” (version 1.22.0) [7]. We utilized the “combinedArray” command to merge the different platforms (850K, EPIC). After filtering and normalization, 265,398 probes remained for subsequent analysis. The 10,000 most variably methylated probes were selected based on the standard deviation to perform unsupervised nonlinear dimension reduction. The resulting distance matrix was used as the input for t-distributed stochastic neighbor embedding analysis (t-SNE; Rtsne package version 0.15). The non-default parameters used were distance=TRUE, perplexity=30, θ=0, and max_iter=2000. A t-SNE plot was generated using the “ggplot2” package (version 3.4.2) for effective visualization. IDAT files were uploaded to versions v11b4 and v12.5 of the online CNS tumor methylation classifier (https://www.molecularneuropathology.org) for classification.
Ethics approval and consent to participate
This study (IRB No: 1905-108-1035) conducted at Seoul National University Hospital (SNUH) has obtained approval from the institutional review board (IRB) and has been carried out in accordance with the ethical principles outlined in the 1964 Declaration of Helsinki and its subsequent revisions. Since this study involves a retrospective review of de-identified electronic medical records, pathology data, and next-generation sequencing (NGS) data using a brain tumor-specific somatic gene panel, the IRB has waived the requirement for informed consent, in compliance with the Korean Bioethics and Safety Act.
RESULTS
Clinical manifestations
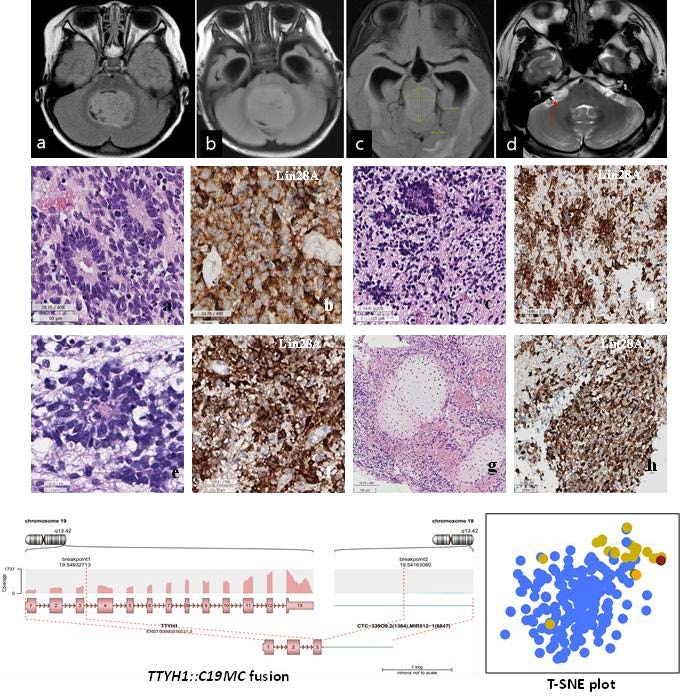
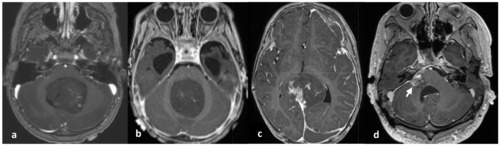
Patient 1 was a previously healthy 4-month-old female who presented with a chief complaint of non-projectile vomiting twice daily for a 2 weeks. Although initially suspected to be acute gastroenteritis, the patient was later admitted when she started experiencing frequent upper eyeball deviation, loss of consciousness, and worsening vomiting. Brain MRI revealed a severe obstructive hydrocephalus, caused by a tumor measuring 4.8 cm in diameter in the 4th ventricle (Fig. 1A). The patient underwent a midline suboccipital craniotomy to remove the tumor.
Patient 2 was a 6-month-old male infant who suddenly developed an inability to hold his head up, accompanied by an enlargement of the head size and projectile vomiting. Brain MRI revealed a mass measuring 6.1×3.7×3.1 cm in the midline posterior fossa of the brain, resulting in obstructive hydrocephalus (Fig. 1B). The patient underwent midline suboccipital craniotomy and tumor removal.
Patient 3 was a 2-year-old female who experienced episodes of waking up with stiffness and shaking in her body. These symptoms worsened over time, occurring during the daytime as well and affecting her sleep and appetite. A brain MRI was performed after the patient exhibited fewer facial wrinkles on the right side when frowning, revealing a mass measuring 3.0×2.4×2.4 cm in the pineal region (Fig. 1C). The patient underwent a craniotomy and tumor removal. Subsequently, the two times more craniotomies were performed and subtotal resection of the recurring tumors in the pineal gland and posterior fossa, respectively, seven months after the initial operation.
Patient 4 was a 30-year-old male who presented with facial palsy, hearing loss, and tinnitus on the right ear that progressively worsened over the course of a month. Brain MRI revealed a well-enhancing nodular mass measuring 2.3 cm at the cerebellopontine angle, initially suspected to be a vestibular schwannoma. The patient initially received a gamma knife surgery, but four months later, he experienced dizziness, nausea, and vomiting. MRI showed a decrease in the size of the mass to 1.4 cm, leading to conservative care. However, four months later, the patient arrived at the emergency room with a sudden onset of severe headache. MRI revealed an increase in the mass size to 4.3×2.5×2.4 cm (Fig. 1D), prompting the patient to undergo a surgical procedure called retromastoid suboccipital craniotomy for tumor removal.
The clinical and pathological characteristics of these four patients are summarized in Table 1 and the initial location and size of ETMR for each patient during the initial imaging study are depicted in Fig. 1.
Patients 1 to 3 were diagnosed with ETMR and received additional chemotherapy using a combination of drugs [Korean society of pediatric neuro-oncology regimen (KSPNO-S-1102A) (Irinotecan, Cisplatin [CDDP], cyclophosphamide [CPM], vincristine [VCR], etoposide [VP16]), along with peripheral blood stem cell mobilization (PBSCM). Case 4 underwent radiotherapy to the right cerebellopontine angle (30 Gy) and one cycle of combination chemotherapy with vincristine, cyclophosphamide and prednisolone. In all four patients, the latest MRI scans revealed the presence of multiple tumor deposits along the leptomeninges, with the hydrocephalus and unfortunately, expired within 1 year. Patient 1 had the shortest follow-up duration of 3.4 months, while patient 3 had the longest at 13.7 months.
The result of the histopathological and immunohistochemical (IHC) study
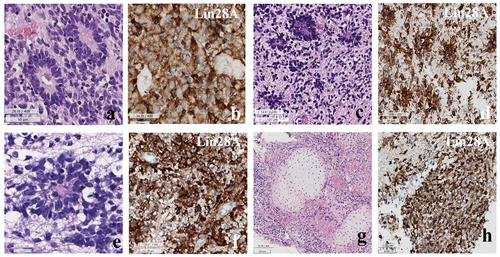
Histopathologically, the tumor from patients 1 through 3 exhibited abundant neutrophils with multilayered rosettes, which is consistent with the characteristic features of this type of tumor (Fig. 2). Patient 4 also displayed these features, but additionally showed chondroid differentiation, which aligns with other known tumors associated with
Immunohistochemical analysis revealed that all cases exhibited positive staining for LIN28A. Additionally, focal positive staining was observed for synaptophysin, neurofilaments (NF), and glial fibrillary acidic protein (GFAP) (Fig. 2). The Ki-67 labeling indices, which reflect the proliferation rate of the tumor cells, were found to be high in all cases, with indices ranging from 52.4% to 89.4% (mean: 70.5%).
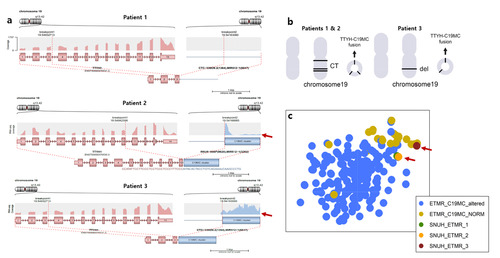
The result of molecular genetic studies
Through NGS tests, it was revealed that patients 1 to 3 exhibited a
The Arriba plots generated from RNA sequencing analysis provided insights into the genomic alterations of patients 1, 2, and 3. Interestingly, patients 1 and 2, who had the
DISCUSSION
ETMRs were first identified as a distinct tumor type in the early 2000s, when Li et al. [8] reported a frequent amplification of the 19q13.41 microRNA amplifications in brain primitive neuroectoderma tumors (PNETs). In 2007, the WHO classified the CNS PNETs subdivided into CNS neuroblastoma/ganglioneuroblastoma, medulloepithelioma (MEPL), ependymoblastoma (EBL), with embryonal tumor with abundant neuropil and true rosettes (ETANTR) discussed as a unique variant [2]. In a landmark paper by Korshunov et al. [9] demonstrated through genome-wide DNA methylation profile analysis of 41 tumors that ENTANTR, MEPL, EBL, and ETANTR, share common LIN28A immunohistochemical positivity and amplification of the chromosome 19q13.42 locus indicating their classification as a single clinicopathological tumor type [2]. Consequently, LIN28A was suggested to screening tool for ETMR [9]. In 2016, the WHO CNS Tumor Classification officially recognized ETMR as a separate tumor entity [10]. However, it was later discovered that LIN28A positivity is not exclusive to ETMR, as it can also be positive in atypical teratoid rhabdoid tumor (ATRT) [1]. Therefore, C19MC amplification testing was also recommended as an additional method to classify a PNET as ETMR [10, 11]. A key study by Lambo et al. [3] revealed that while C19MC amplification is observed in 90% of ETMR cases, the next most common is
We have conducted a study following the established methods to confirm the LIN28A IHC positivity in all 4 of our patients with ETMR. Moreover, we molecularly verified the presence of C19MC amplification and
The prognosis for ETMR is dismal, primarily due to its epidemiology and rarity. Since the majority of cases occur in the children below the age of 2, with very few cases reported in older children, the use of high-dose chemotherapy is limited by its associated toxicities [13, 14]. Radiation therapy can be effective but it accompanied by several severe side effects [14]. Proton therapy has been suggested as an alternative to reduce side effects compared to conventional radiation therapy [15]. However, targeted therapies may offer a promising approach to minimize side effects. Targeting R-loops with topoisomerase and PARP inhibitors has been proposed as a potential strategy [13]. DNMT3B, which is active only during the early first weeks of neural tube development, has been suggested as a candidate for future therapies even prior to the inclusion of ETMR in the current WHO Classification of Tumors [16]. Furthermore, pathways such as MIR17HG, WNT SHH, or mTOR pathways could be potential targets downstream from the known C19MC amplification and
Patient 4, an adult male aged 30, presented a unique case of ETMR with a
Genetic counseling is strongly recommended for patients with
In the context of
In conclusion, we have presented three cases of ETMR characterized by typical C19MC amplification, along with a
ACKNOWLEDGEMENTS
This study was supported by a grant of the Korea Health Technology R&D Project through the Korea Health Industry Development Institute (KHIDI), funded by the Ministry of Health & Welfare, Republic of Korea (Grant number: HI14C1277) and the Institute for Information & Communications Technology Promotion (IITP) grant funded by the Korean government (MSIP) (No.2019-0567).
AUTHOR CONTRIBUTIONS
S.-H.P. designed and supervised the study and E.E.K. wrote the entire manuscript. K.L. analyzed the methylation data and wrote the manuscript. S.-H.P. and E.E.K. reviewed histopathology slides and anonymized molecular data for qualitative analysis. J.-H. P. and M.-S.K. operated and treated the patients, and provided clinical data. H.Y. analyzed the NGS data derived from the NGS studies using a customized brain tumor targeted gene panel. All authors have reviewed and edited the final manuscript. All materials had been obtained for the electronic medical record of the patients, which were anonymized and retrospectively reviewed. No extra-human materials were obtained from the patients for this study. Under the Korean Bioethics and Safety Act, additional consent to publish was waived.
CONFLICT OF INTEREST
The authors declare no competing interests.
Figures
{kind=link}
{kind=link}
{kind=link}
Tables
Clinicopathological features of three patients with
| Pt | Gender | Age | NGS results | LIN28A IHC | Site | OP | Adjuvant | F-U dur | Last MRI |
|---|---|---|---|---|---|---|---|---|---|
| 1 | F | 4 months | C19MC amp (×7) | + | 4th ventricle | NTR | CTx | 3.4 months | Multiple seeding, hydrocephalus |
| 2 | M | 9 months | C19MC amp (×5) | + | Midline posterior fossa | STR | CTx | 9.8 months | Multiple seeding, hydrocephalus |
| 3 | F | 2 years | C19MC amp (×7) | + | Pineal gland | STR | CTx | 13.7 months | Multiple seeding, hydrocephalus |
| 4 | M | 30 years | + | Rt. CPA | GTR | RT to Rt. CPA (30 Gy)+CTx (VCP #1) | 8.0 months | Multiple seeding, hydrocephalus |
References
- Rao S, Rajeswarie RT, Chickabasaviah Yasha T, Nandeesh BN, Arivazhagan A, Santosh V (2017) LIN28A, a sensitive immunohistochemical marker for embryonal tumor with multilayered rosettes (ETMR), is also positive in a subset of atypical teratoid/rhabdoid tumor (AT/RT). Childs Nerv Syst 33:1953-1959


- Korshunov A, Sturm D, Ryzhova M, Hovestadt V, Gessi M, Jones DT, Remke M, Northcott P, Perry A, Picard D, Rosenblum M, Antonelli M, Aronica E, Schüller U, Hasselblatt M, Woehrer A, Zheludkova O, Kumirova E, Puget S, Taylor MD, Giangaspero F, Peter Collins V, von Deimling A, Lichter P, Huang A, Pietsch T, Pfister SM, Kool M (2014) Embryonal tumor with abundant neuropil and true rosettes (ETANTR), ependymoblastoma, and medulloepithelioma share molecular similarity and comprise a single clinicopathological entity. Acta Neuropathol 128:279-289

- Lambo S, Gröbner SN, Rausch T, Waszak SM, Schmidt C, Gorthi A, Romero JC, Mauermann M, Brabetz S, Krausert S, Buchhalter I, Koster J, Zwijnenburg DA, Sill M, Hübner JM, Mack N, Schwalm B, Ryzhova M, Hovestadt V, Papillon-Cavanagh S, Chan JA, Landgraf P, Ho B, Milde T, Witt O, Ecker J, Sahm F, Sumerauer D, Ellison DW, Orr BA, Darabi A, Haberler C, Figarella-Branger D, Wesseling P, Schittenhelm J, Remke M, Taylor MD, Gil-da-Costa MJ, Łastowska M, Grajkowska W, Hasselblatt M, Hauser P, Pietsch T, Uro-Coste E, Bourdeaut F, Masliah-Planchon J, Rigau V, Alexandrescu S, Wolf S, Li XN, Schüller U, Snuderl M, Karajannis MA, Giangaspero F, Jabado N, von Deimling A, Jones DTW, Korbel JO, von Hoff K, Lichter P, Huang A, Bishop AJR, Pfister SM, Korshunov A, Kool M (2019) The molecular landscape of ETMR at diagnosis and relapse. Nature 576:274-280
- Uro-Coste E, Masliah-Planchon J, Siegfried A, Blanluet M, Lambo S, Kool M, Roujeau T, Boetto S, Palenzuela G, Bertozzi AI, Gambart M, Coupier I, Oliver-Petit I, Golmard L, Julia S, Savagner F, Mohand-Oumoussa B, Tauziede-Espariat A, Delisle MB, Figarella-Branger D, Bourdeaut F, Rigau V (2019) ETMR-like infantile cerebellar embryonal tumors in the extended morphologic spectrum of DICER1-related tumors. Acta Neuropathol 137:175-177
- Lee K, Kim SI, Kim EE, Shim YM, Won JK, Park CK, Choi SH, Yun H, Lee H, Park SH (2023) Genomic profiles of IDH-mutant gliomas: MYCN-amplified IDH-mutant astrocytoma had the worst prognosis. Sci Rep 13:6761
- Capper D, Jones DTW, Sill M, Hovestadt V, Schrimpf D, Sturm D, Koelsche C, Sahm F, Chavez L, Reuss DE, Kratz A, Wefers AK, Huang K, Pajtler KW, Schweizer L, Stichel D, Olar A, Engel NW, Lindenberg K, Harter PN, Braczynski AK, Plate KH, Dohmen H, Garvalov BK, Coras R, Hölsken A, Hewer E, Bewerunge-Hudler M, Schick M, Fischer R, Beschorner R, Schittenhelm J, Staszewski O, Wani K, Varlet P, Pages M, Temming P, Lohmann D, Selt F, Witt H, Milde T, Witt O, Aronica E, Giangaspero F, Rushing E, Scheurlen W, Geisenberger C, Rodriguez FJ, Becker A, Preusser M, Haberler C, Bjerkvig R, Cryan J, Farrell M, Deckert M, Hench J, Frank S, Serrano J, Kannan K, Tsirigos A, Brück W, Hofer S, Brehmer S, Seiz-Rosenhagen M, Hänggi D, Hans V, Rozsnoki S, Hansford JR, Kohlhof P, Kristensen BW, Lechner M, Lopes B, Mawrin C, Ketter R, Kulozik A, Khatib Z, Heppner F, Koch A, Jouvet A, Keohane C, Mühleisen H, Mueller W, Pohl U, Prinz M, Benner A, Zapatka M, Gottardo NG, Driever PH, Kramm CM, Müller HL, Rutkowski S, von Hoff K, Frühwald MC, Gnekow A, Fleischhack G, Tippelt S, Calaminus G, Monoranu CM, Perry A, Jones C, Jacques TS, Radlwimmer B, Gessi M, Pietsch T, Schramm J, Schackert G, Westphal M, Reifenberger G, Wesseling P, Weller M, Collins VP, Blümcke I, Bendszus M, Debus J, Huang A, Jabado N, Northcott PA, Paulus W, Gajjar A, Robinson GW, Taylor MD, Jaunmuktane Z, Ryzhova M, Platten M, Unterberg A, Wick W, Karajannis MA, Mittelbronn M, Acker T, Hartmann C, Aldape K, Schüller U, Buslei R, Lichter P, Kool M, Herold-Mende C, Ellison DW, Hasselblatt M, Snuderl M, Brandner S, Korshunov A, von Deimling A, Pfister SM (2018) DNA methylation-based classification of central nervous system tumours. Nature 555:469-474
- Maksimovic J, Phipson B, Oshlack A (2016) A cross-package Bioconductor workflow for analysing methylation array data. F1000Res 5:1281
- Li M, Lee KF, Lu Y, Clarke I, Shih D, Eberhart C, Collins VP, Van Meter T, Picard D, Zhou L, Boutros PC, Modena P, Liang ML, Scherer SW, Bouffet E, Rutka JT, Pomeroy SL, Lau CC, Taylor MD, Gajjar A, Dirks PB, Hawkins CE, Huang A (2009) Frequent amplification of a chr19q13.41 microRNA polycistron in aggressive primitive neuroectodermal brain tumors. Cancer Cell 16:533-546
- Korshunov A, Ryzhova M, Jones DT, Northcott PA, van Sluis P, Volckmann R, Koster J, Versteeg R, Cowdrey C, Perry A, Picard D, Rosenblum M, Giangaspero F, Aronica E, Schüller U, Hasselblatt M, Collins VP, von Deimling A, Lichter P, Huang A, Pfister SM, Kool M (2012) LIN28A immunoreactivity is a potent diagnostic marker of embryonal tumor with multilayered rosettes (ETMR). Acta Neuropathol 124:875-881
- World Health Organization (WHO) (2021) WHO classification of tumours: central nervous system tumours. 5th ed. International Agency for Research on Cancer Publications, Lyon
- Wesseling P (2014) Embryonal tumor with multilayered rosettes (ETMR): signed, sealed, delivered …. Acta Neuropathol 128:305-308
- von Hoff K, Haberler C, Schmitt-Hoffner F, Schepke E, de Rojas T, Jacobs S, Zapotocky M, Sumerauer D, Perek-Polnik M, Dufour C, van Vuurden D, Slavc I, Gojo J, Pickles JC, Gerber NU, Massimino M, Gil-da-Costa MJ, Garami M, Kumirova E, Sehested A, Scheie D, Cruz O, Moreno L, Cho J, Zeller B, Bovenschen N, Grotzer M, Alderete D, Snuderl M, Zheludkova O, Golanov A, Okonechnikov K, Mynarek M, Juhnke BO, Rutkowski S, Schüller U, Pizer B, von Zezschwitz B, Kwiecien R, Wechsung M, Konietschke F, Hwang EI, Sturm D, Pfister SM, von Deimling A, Rushing EJ, Ryzhova M, Hauser P, Łastowska M, Wesseling P, Giangaspero F, Hawkins C, Figarella-Branger D, Eberhart C, Burger P, Gessi M, Korshunov A, Jacques TS, Capper D, Pietsch T, Kool M (2021) Therapeutic implications of improved molecular diagnostics for rare CNS embryonal tumor entities: results of an international, retrospective study. Neuro Oncol 23:1597-1611
- Lambo S, Gröbner SN, Rausch T, Waszak SM, Schmidt C, Gorthi A, Romero JC, Mauermann M, Brabetz S, Krausert S, Buchhalter I, Koster J, Zwijnenburg DA, Sill M, Hübner JM, Mack N, Schwalm B, Ryzhova M, Hovestadt V, Papillon-Cavanagh S, Chan JA, Landgraf P, Ho B, Milde T, Witt O, Ecker J, Sahm F, Sumerauer D, Ellison DW, Orr BA, Darabi A, Haberler C, Figarella-Branger D, Wesseling P, Schittenhelm J, Remke M, Taylor MD, Gil-da-Costa MJ, Łastowska M, Grajkowska W, Hasselblatt M, Hauser P, Pietsch T, Uro-Coste E, Bourdeaut F, Masliah-Planchon J, Rigau V, Alexandrescu S, Wolf S, Li XN, Schüller U, Snuderl M, Karajannis MA, Giangaspero F, Jabado N, von Deimling A, Jones DTW, Korbel JO, von Hoff K, Lichter P, Huang A, Bishop AJR, Pfister SM, Korshunov A, Kool M (2019) The molecular landscape of ETMR at diagnosis and relapse. Nature 576:274-280
- Bandopadhayay P, Chi SN (2022) The challenges in treating embryonal tumors with multilayered rosettes (ETMR) and other infant brain tumors. Neuro Oncol 24:138-140
- Jaramillo S, Grosshans DR, Philip N, Varan A, Akyüz C, McAleer MF, Mahajan A, McGovern SL (2018) Radiation for ETMR: literature review and case series of patients treated with proton therapy. Clin Transl Radiat Oncol 15:31-37
- Kleinman CL, Gerges N, Papillon-Cavanagh S, Sin-Chan P, Pramatarova A, Quang DA, Adoue V, Busche S, Caron M, Djambazian H, Bemmo A, Fontebasso AM, Spence T, Schwartzentruber J, Albrecht S, Hauser P, Garami M, Klekner A, Bognar L, Montes JL, Staffa A, Montpetit A, Berube P, Zakrzewska M, Zakrzewski K, Liberski PP, Dong Z, Siegel PM, Duchaine T, Perotti C, Fleming A, Faury D, Remke M, Gallo M, Dirks P, Taylor MD, Sladek R, Pastinen T, Chan JA, Huang A, Majewski J, Jabado N (2014) Fusion of TTYH1 with the C19MC microRNA cluster drives expression of a brain-specific DNMT3B isoform in the embryonal brain tumor ETMR. Nat Genet 46:39-44
- Apellaniz-Ruiz M, Sabbaghian N, Chong AL, de Kock L, Cetinkaya S, Bayramoğlu E, Dinjens WNM, McCluggage WG, Wagner A, Yilmaz AA, Foulkes WD (2023) Reclassification of two germline DICER1 splicing variants leads to DICER1 syndrome diagnosis. Fam Cancer 22:487-493
- Klein S, Lee H, Ghahremani S, Kempert P, Ischander M, Teitell MA, Nelson SF, Martinez-Agosto JA (2014) Expanding the phenotype of mutations in DICER1: mosaic missense mutations in the RNase IIIb domain of DICER1 cause GLOW syndrome. J Med Genet 51:294-302
- Foulkes WD, Priest JR, Duchaine TF (2014) DICER1: mutations, microRNAs and mechanisms. Nat Rev Cancer 14:662-672