Articles

Article Tools
 View Full Text View Full Text |
 Abstract Abstract |
 Article as PDF Article as PDF |
 Print this Article Print this Article |
 Pubmed Pubmed |
 PMC PMC |
 PubReader PubReader |
 Export to Citation Export to Citation |
 Email Alerts Email Alerts |
 Open Access Open Access |
Supplementary
Share this article on :



Stats or Metrics
Article
Original Article
Exp Neurobiol 2019; 28(4): 474-484
Published online August 31, 2019
https://doi.org/10.5607/en.2019.28.4.474
© The Korean Society for Brain and Neural Sciences
Distinct Topographical Patterns of Spike-Wave Discharge in Transgenic and Pharmacologically Induced Absence Seizure Models
Soojung Lee1, Eunjin Hwang2, Mina Lee2,3 and Jee Hyun Choi2,3*
1Department of Oral Physiology, Faculty of Dentistry, Kyung Hee University, Seoul 02447, 2Center for Neuroscience, Korea Institute of Science and Technology, Seoul 02792, 3Department of Neuroscience, University of Science and Technology, Daejeon 34113, Korea
Correspondence to: *To whom correspondence should be addressed.
TEL: 82-2-958-6952, FAX: 82-2-958-6737
e-mail: jeechoi@kist.re.kr
This is an Open Access article distributed under the terms of the Creative Commons Attribution Non-Commercial License(http://creativecommons.org/licenses/by-nc/4.0) which permits unrestricted non-commercial use, distribution, andreproduction in any medium, provided the original work is properly cited.
Abstract
Absence seizures (AS) are generalized non-convulsive seizures characterized by a brief loss of consciousness and spike-and-wave discharges (SWD) in an electroencephalogram (EEG). A number of animal models have been developed to explain the mechanisms of AS, and thalamo-cortical networks are considered to be involved. However, the cortical foci have not been well described in mouse models of AS. This study aims to use a high density EEG in pathophysiologically different AS models to compare the spatiotemporal patterns of SWDs. We used two AS models: a pharmacologically induced model (gamma-hydroxybutyric acid, GHB model) and a transgenic model (phospholipase beta4 knock-out, PLCβ4 model). The occurrences of SWDs were confirmed by thalamic recordings. The topographical analysis of SWDs showed that the onset and propagation patterns were markedly distinguishable between the two models. In the PLCβ4 model, the foci were located within the somatosensory cortex followed by propagation to the frontal cortex, whereas in the GHB model, a majority of SWDs was initiated in the prefrontal cortex followed by propagation to the posterior cortex. In addition, in the GHB model, foci were also observed in other cortical areas. This observation indicates that different cortical networks are involved in the generation of SWDs across the two models.
Graphical Abstract
Keywords: Electroencephalogram, Mice, Absence seizure, Spike-wave discharge
INTRODUCTION
Absence seizures (AS) are generalized non-convulsive seizures, accompanied by a brief loss of consciousness with bilateral spike-and-wave discharges (SWD) at a frequency of 2.5~4 Hz in a human electroencephalogram (EEG) and are generally associated with mild impairments in cognition [1, 2]. A number of patho-physiological animal models have been developed to explain the mechanisms of AS generation, because human studies can only provide limited information using non-invasive whole brain techniques. Multiple lines of evidence strongly suggested that abnormally hypersynchronized oscillations within the thalamocortical circuitry are the primary underlying mechanisms for SWD generation [3, 4]. However, recent developments in genetic rodent models have effectively argued that the cortex may play the primary role in the generation of SWDs [5–8]. In clinic, it is known that absence seizures are predominantly genetic in etiology. While the brain activities recorded in the EEG are the standard exam for the diagnosis of absence seizure, significant differences in the SWD patterns were found leading a difficulty in building heuristic treatment or therapeutic strategies. A direct cross-specicies comparison of cortical topographies of SWD between human and mouse EEG is expected to correlate the EEG topographies with causes at the neuronal levels but has been limited due to lack of spatial information of mouse EEG.
Multiple lines of evidence have suggested that SWDs are not primarily generalized despite its definition, and rather involve particular neuronal networks with focal cortical onset [9–12]. In human magnetoencephalography (MEG), the distribution of SWDs appeared to involve a prefrontal-insular-thalamic network [13]. EEG with functional MRI (EEG-fMRI) has also identified a common network of structures involved in AS generation [13], comprising the thalamus, midline and lateral parietal cortex (a subset of default mode network, DMN), and striatum (predominantly caudate nuclei) [1]. These networks are known to be a large-scale network of heteromodal associative parietal and frontal cortical areas, engaged in internalized cognitive activity, such as random reflective thoughts and free associations of ideas and memories [1, 14, 15]. Furthermore, pharmacological or genetic studies using animal models demonstrated that thalamic complex neurons have been strongly suggested to be important for SWD generation [16–20], but the spatiotemporal aspects of thalamocortical (TC) interactions that underlie seizure manifestations have not yet been thoroughly explained. The physiological role of TC networks in the maintenance of the sleep-wake cycle in relation to sleep spindles is well-established [21]. However, its relevance to SWDs has been questioned, as ASs predominantly occur during wakeful and drowsy states but rarely in NREM sleep [22–24]. The dissimilarity between sleep spindles and SWDs was also noted but poorly answered in genetic rodent AS models [24–26], challenging the longstanding concept of initiation of bilateral epileptic discharges from sleep spindle within the thalamic complex in network models.
Our aim in this study was to characterize the topographical patterns of SWD in mouse model. Compared to widely explored rodent genetic models (WAG/Rij and GAERS) – which were instrumental in establishing much of what we know about the circuitry and molecular mechanisms underlying the epileptogenic process and effectiveness of anticonvulsant drugs – monogenic mutations (Stargazer, tottering and lethargic) or pharmacological models (gamma-butyrolactone and pentylenetetrazole) give insights into the mechanism of epileptogenesis with a level of precision to a singular pathogenic molecular defect, as only genes related to ion channels have so far been linked to absence phenotypes [27–31]. As the gamma-butyrolactone induced absence seizure model (GHB model) and the phospholipase beta4 knock out mice model (PLCβ4 model) are known to heavily target TC circuits, we sought to determine the relative roles of the different components of TC circuits using the whole cortical activity, simultaneously monitored by a high-density EEG (hd-EEG) [32] and local field potential (LFP) electrodes. By applying this functional brain mapping technique, our results from the GHB model and the PLCβ4 model reveal different spatiotemporal patterns of SWDs, as well as different relationships between the cortex and thalamus during paroxysmal epileptic seizures. In the two AS models, we also consider the functional significance of cortical networks engaged in seizure initiation and manifestations during epileptic seizure generation within the brain network.
MATERIALS AND METHODS
Ethics statement
All surgical and experimental procedures were followed by Korean Animal and Plant Quarantine Agency Publication No. 12512, partial amendment 2014, conforming to NIH guidelines (NIH Publication No. 86-23, revised 1985). All the procedures were approved by Korea Institute of Science and Technology and Institutional Animal Care and Use Committee of Korea Institute of Science and Technology (AP-2014L7002).
Absence seizure models
For the experiments, the PLCβ4 knock-out mice were generated according to a previous report, derived from mating heterozygous mice of 129S4/SvJae and C57BL/6J genetic backgrounds [33]. The PLCβ4 is highly expressed in thalamus, isocortex, hypothalamus, brainstem, and cerebellum according to ISH data in Allen Brain Atlas (
Implantation of EEG and LFP electrodes
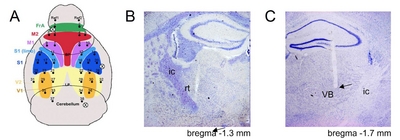
The surgery and experiments were performed in 10- to 14-week old male mice. Animals were anesthetized with a ketamine and xylazine cocktail (120 and 6 mg/kg, respectively) by intraperitoneal injection, and positioned in a stereotaxic apparatus (Model 900, David Kopf Instruments, Tujunga, CA, USA). Whenever whisker movements were observed during the surgery, supplemental anesthetic (a third of the original dose) was given. After skull exposition, the high-density EEG microarray was placed on the skull as previously described in detail [32] with video demonstration [36]. In addition to the microarray, two Teflon-coated tungsten electrodes (φ=115 μm, A-M systems, Everett, WA, USA) were implanted into the ventroposterior thalamus (VP, AP: −1.6 mm, ML: −1.7 mm, DV: 3.4~3.7 mm) and the thalamic reticular nuclei (nRT, AP: −0.6 mm, ML: 1.3 mm, DV: 3.0~3.2 mm). Two microscrews (chrome-plated stainless steel, Asia Bolt, Seoul, Korea) were fixed onto the skull above the right cerebellum (AP: −5.7 mm, ML: 1.7 mm) and the right olfactory bulb (AP: 4.5 mm, ML: 1.5 mm) for reference and ground electrodes for LFP electrodes respectively. The electrodes were secured to the skull with dental acrylic cement (Vertex-Dental, Vertex Self-Curing, Zeist, The Netherlands). The incised skin was sutured and antibiotic cream was applied. After surgery, the mice were allowed to recover in individual cages for at least a week. The montage of high density EEG and the histological confirmation are in Fig. 1A. The locations of LFP tips were confirmed after experiments (Fig. 1B and 1C) and wrong positions were excluded in further analysis.
EEG recording and movement monitoring
The high-density EEG and thalamic LFP were simultaneously recorded using a SynAmp2 amplifier (Neuroscan Inc., El Paso, TX, USA). All signals were digitized with 1 kHz sampling rate and band-pass filtered from 0.1 to 100 Hz. The impedance of microarray channels was maintained < 100 kΩ (at 30 Hz test frequency). All recordings were performed in a clean beaker to prevent any excessive explorative behaviors, and videos were recorded to observe animal behaviors. Recordings were performed for 1 and 2 hours in the case of the GHB model mice and the PLCβ4 model mice, respectively.
Detection of SWDs
Before detection of SWDs, visual inspection was performed on the hdEEG and the periods of contaminated signals were excluded prior to applying the algorithm. SWDs in each channel were detected automatically with the following steps: 1) we normalized the signals with average power in the frequency range of 90~100 Hz to set the impedance levels of electrodes to a similar level, then a band-pass filter with cutoff frequency of 1 and 30 Hz was applied with a 8th order zero-phase delay Butterworth filter. 2) The standard deviation of the signals during the quiescent awake baseline period was calculated and used as the threshold for event detection. 3) In individual channels, all peaks above the threshold were detected using peak detection algorithm. 4). The periods with a repetition of more than two peaks with inter-peak distance in the range of 100~500 ms were selected, and those periods longer than 0.5 s were classified as potential SWD events. 5) The coefficients of multiple correlation,
Determining existence of waxing and waning patterns in SWD
To investigate whether the SWD presents a waxing and waning morphology with round peaks and valley, we applied the linear and quadratic regression models on the sequence of peak-to-peak amplitude in the spikes and then compared the goodness-of-fit between two models. We used
Spectral analysis and topography
The high-density EEG data was analyzed with a custom-made MATLAB (Mathworks, Inc. Natick, MA, USA) program. The channels with contact impedance higher than 1 MΩ were excluded from the analysis. Spectral analysis was performed by Welch’s method with a 1-s Hanning window and a 0.25-Hz frequency bin size. Mean power of peak frequency (3~5 Hz and 6~8 Hz in the GHB model and the PLCβ4 model, respectively) was presented as topography. The topographical mapping was represented by a color map on the mouse brain surface which was rendered by a ‘spm_surf’ function in SPM8 (Wellcome Trust Centre for Neuro-imaging, UCL, London, UK) using the mouse magnetic resonance microscopy atlas (downloadable in
Histological analysis
For histology, the post-recorded mice were deeply anesthetized with 2% avertin and transcardially perfused with 200~300 ml of 0.9% NaCl followed by 200 ml of 4% paraformaldehyde in 1 X PBS. Perfused brains were removed and fixed overnight in the same fixative at 4°C. Brains were then frozen in freezing media and coronal sections (30 μm) were cut by cryostat HM525 (Thermo Scientific Inc. Microm, Walldorf, Germany). The brain slices were collected in tissue storage buffer and images were visualized with BX50 (Olympus Cor., Tokyo, Japan) to verify electrode positions.
Statistical analysis
For identifying the channels with significantly increased power compared to baseline fluctuation, Student’s paired t-test was employed. For comparing SWD variables (e.g. event duration, and the duration and amplitude of spikes) between the two models, a non-parametric hypothesis test, Kolmogorov-Smirnov test was used. The difference was considered to be significant if p<0.05.
Data availability
The raw data that support the findings of this study are available from the corresponding author upon reasonable request but restrictions apply to the availability of these data publicly.
RESULTS
Spike and wave discharge patterns
Spike-wave-discharge bursts of typical absence seizures were observed in high-density EEG data across a total of 43 SWDs in 5 of the GHB model mice and a total of 47 SWDs in 6 of the PLCβ4 model mice. The mean occurrence rate of seizures in the GHB model was 2.5±2.6 per min. Typically, SWDs started to occur 5 to 10 min after the injection of gamma-butyrolactone (GBL, pro-drug of GHB), and only the episodic SWDs were counted for data analysis. In the PLCβ4 model, all the SWD events have similar waveform characteristics in terms of spike and wave patterns. On the other hand, the SWDs observed in GHB model have relatively dissimilar waveform characteristics compared to PLCβ4 model. The occurrence rate was 4.1±2.4 per permin in the awake PLCβ4 model, which was not significantly different from the GHB model (Two-sample Komogorov-Smirnov test, p-value=3.98E-21). The mean durations of SWDs were 1.48±0.61 sec and 1.13±0.30 sec in the GHB model and the PLCβ4 model respectively (Two-sample Kolmogorov-Smirnov test, p-value=1.88E-5). We double-checked the thalamic LFPs to avoid any false detection of swing noise produced by abrupt moves in the interface between wires and connector board of microarray. The raw traces of high-density EEG and LFP signals containing SWDs are exemplified in Fig. 2C and 2D.
Wave patterns of SWDs were clearly distinguishable between the two models
First, the PLCβ4 model had sharper spikes with a positive polarity compared to the spikes observed in the GHB model. The spike width (i.e. the time interval between a pair of successive negative and positive peaks) in the GHB model was 82.1±15.0 msec, whereas it was 25.6±5.4 msec in the PLCβ4 model (Two-sample Kolmogorov-Smirnov test, p-value=1.80E-14). Conversely, the overall peak amplitudes (i.e. the voltage difference between the negative and positive peaks) in the two models were not significantly different: 255±427 μV in the GHB model and 208±258 μV in the PLCβ4 model (Two-sample Kolmogorov-Smirnov test, p-value=0.6851).
Second, the traces of SWDs in the GHB model mice tended to have a waxing and waning rhythmic pattern [37, 38] that is attributed to a progressive entrainment of spikes into the oscillation, followed by a progressive desynchronization [39]. A comprehensive analysis based on nonlinear regression of the quadratic curve revealed that 35 out of 43 SWD events in the GHB model showed waxing and waning patterns, although not in all of the channels. A total of 24.2±19.8% of SWD-detected channels showed waxing and waning SWD patterns. Conversely, in the PLCβ4 model, 34 out of 47 SWDs events showed waxing and waning patterns in 2.0±4.1% of SWD-detected channels.
Third, the mean frequency of SWDs, calculated by the inverse of peak intervals, was significantly different between the two mouse models: the frequency in the GHB model was 4.16±1.12 Hz, whereas in the PLCβ4 model it was 7.37±0.74 Hz. Fig. 1E and 1F showed the representative spectrogram of EEG in the primary somatosensory cortex.
Topographical analysis of power and spike amplitude
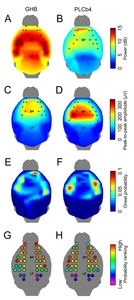
High-density EEG data in a large area of the mouse cortex was examined in a topographic presentation on the mouse brain surface. First, the spatial distribution of spectral power in the seizure bands (3~5 Hz and 6~8 Hz in the GHB and PLCβ4 models respectively) were plotted over the cortex (Fig. 3A and 3B). The EEG channels with significantly increased spectral power compared to the baseline fluctuations were marked as black dots. In both models, the significantly altered channels were located in the prefrontal, motor, and anterior side of the somatosensory cortex, while more temporal channels were affected in the PLCβ4 model. Generally, the spectral power was larger in the GHB model compared to the PLCβ4 model. By applying the Fourier transform, the spectral power measures the signal levels of waves with wavelength of 200~333 ms and 125~167 ms for the GHB and the PLCβ4 models respectively. The waves with these wavelength ranges were the discharges followed by spikes, suggesting that the wave discharges followed by spikes were stronger in the GHB model than in the PLCβ4 model.
In order to estimate the magnitude of the spikes, we calculated the voltage difference between the negative peak and the following positive peak of the spikes, and depicted their topography in Fig. 3C and 3D. In the GHB model, the the highest amplitude spikes were found in the medial frontal cortex, while the lateral parietal cortex presented the largest amplitude spike in the PLCβ4 model. The channel with the largest spike amplitude is on top of the somatosensory cortex (see the EEG montage in
Spatial distributions of seizure onset
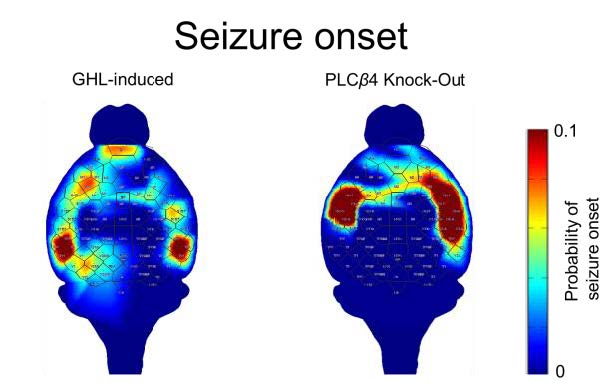
The onset patterns indicating the initiation of SWDs were investigated by pinpointing the temporal point of the first peak in each EEG channel after the moment of crossing a preset threshold. Fig. 2E and 2F displays the probability map of the seizure onset. Although multiple foci were observed in both models, the somatosensory regions were highly likely to be the initiating regions of SWDs in the PLCβ4 model, whereas the onset patterns were more scattered in the GHB model. The onset probability ranking of each channel showed that most SWDs were initiated within the somatosensory cortex in the PLCβ4 model, whereas the SWDs in the were initiated most likely in the prefrontal cortex in the GHB model (Fig. 3G and 3H). In addition, foci were also observed in other cortical areas in the GHB model.
DISCUSSION
Spike and wave discharge patterns
In this paper, we studied the topographical patterns of spike-wave discharges in transgenic and pharmacologically induced absence seizure models. The origin of the SWDs remains controversial. Some studies propose the cortical onset, and others the subcortical one (diencephalon), both based on animal and human data [40]. While “thalamic clock” theories support the hypothesis that sleep-like spindles give rise to the absence-related SWDs that originate in the subcortical structures, particularly in the thalamic complex, “cortical focus” theories suggest that leading spikes may originate around the cortical somatosensory areas and the thalamus provides a resonant circuitry to amplify and sustain the discharges [5, 41, 42]. “Cortical reticular” theory, which is still widely accepted, suggests that hyperexcitable cortex transforms normal sleep spindles into the paroxysmal oscillation of the SWDbut the relative contributions of the cortex and the thalamus and their interactions are still a matter of debate [42, 43]. Together with lesion studies of the reticular thalamic nucleus [26], various findings in thalamocortical neurons in absence models – such as the upregulations of the various types of Ca2+ channels, anomalies of Ih, and the lack of some of the GABA-subunits in the reticular thalamic neurons – imply that the thalamic complex may still be an important site for the pathophysiology of SWD generation [17, 19, 30, 44, 45]. PLCβ4, a downstream signaling molecule of type 1 metabotropic glutamate receptors, is highly expressed in TC neurons but far less in nRT and the cortex in whole brain quantification of knock-out model [46]. Global deletion of PLCβ4 in the whole brain caused changes in the intrinsic properties of TC neurons, SWD generation in thalamocortical networks, and absence-like behavioral changes [33, 47]. The excitatory synaptic inputs in TC and nRT neurons from the cortex through the corticothalamic feedback loop would increase in the PLCβ4 model [48], due to the decreased tonic firing of TC neurons [49]. Given the scarcity of rebound T-channel bursts in TC neurons in vitro and in intact animal studies [50, 51], increased T-channel bursts in these neurons may not be relevant to SWD generation in the PLCβ4 model, whereas the bursts of nRT neurons play a key role in SWDs development and their cycles, due to their feedforward inhibition of TC neurons [52, 53]. The combination of increased excitatory inputs to TC and nRT from the cortex on the one hand, and the resultant enhanced bursts in nRT on the other, may contribute to the generation of the phase-locked paroxysmal oscillations and the more rapid synchronous spreading of SWDs in the cortex.
Considering that the oscillations on a large-scale level reflect the membrane resonant states on a cellular level, the different frequencies in SWDs, i.e. spike width differences, imply that the two models have different underlying mechanisms for driving the resonant oscillations. Strong similarities in the firing dynamics and neuronal interactions in corticothalamo-cortical circuits between the two models suggest that similar mechanisms may underlie SWD generation in the thalamic complex. The complex dynamics of TC and nRT neuron firings were similarly demonstrated in the rodent GAERS and the GHB models recently, where a selective blockade of T-channels in NRT and cortical neurons decreased ASs, emphasizing the importance of nRT in SWD generation [54]. The injection of GBL in PLCβ4 knock-out mice reportedly induced slower SWDs (2~5 Hz) in the frontal cortex and the ventrobasal thalamic complex – the same frequency as observed in the GHB model [33]. This intervention masked faster kinetics of innate resonance (approx. 7 Hz) in the PLCβ4 model, rather than generating two different frequency oscillations. This finding implies that the SWDs from both the GHB model and the GBL injected PLCβ4 knock-out mice, governed by the slow kinetics of GABAB receptors, share certain common networks which underlie SWD generation [18]. Targeting extrasynatpic GABAA receptors by the GHB in TC and layer IV cortical neurons may also lead to slower kinetics of SWDs compared to the faster kinetics of SWDs in genetic models [55]. However, it was not known whether these innate resonant SWDs in the PLCβ4 model were also attenuated in other brain regions. It should be emphasized that GBL only induced ASs in human subjects with a history of epileptic seizures, but never without such a history. That the humans without a history of seizures reportedly retained minimal consciousness implies that the GHB does not completely affect consciousness networks in humans. It would be interesting to compare an epileptogenic network in GBL treated AS patients with SWD generation networks in the rodent GHB model.
Spatial distribution of seizure onset
Prior research suggested that cortical initiation sites were localized in layers V~VI of the somatosensory cortex or the perioral region of the somatosensory cortex in genetic AS rodent models [2], but few studies have explored SWD’s spatial distribution in the GHB model [29, 56]. A recent study suggested that the SWDs are focal in onset, evolving from the somatosensory cortex [32] and spreading to other cortical areas and the thalamus with a reverberating pattern. In this study, the power of the SWDs in the posterior parietal or occipital lobes did not increase significantly during SWDs.
Evaluating cortical high-density EEG and thalamic LFP signals of both models in this study, we found that most SWDs were initiated within the somatosensory cortex in the PLCβ4 model, whereas in the GHB model the initiation of SWDs was dispersed throughout the cortex, although initiation sites were distinctly localized within the cortex. We did not observe that SWD initiation is linked to prior sleep spindle generation. It seemed nRT was not affected by both the GHB and the deletion of PLCβ4. Topographical spatiotemporal patterns of SWD were clearly discernible between the two models, even though multiple foci were observed in both models: widely spreading SWDs (GHB model) vs. relatively focal SWDs (PLCβ4 model). In consideration of the dispersive perfusion of the drug into the brain and the global distribution of GABAB and GHB receptors [57] throughout the brain, it is interesting to note that the power of the SWDs in the GHB model is mostly confined to the frontal regions rather than being generalized, and that the onset patterns were more scattered than the PLCβ4 model. Conversely, in the case of the PLCβ4 model, relatively confined cortical areas engaging SWD generation strongly indicates that the particular cortical networks are involved in the generation of spontaneous SWDs, because very low levels of expression of PLCβ4 in the cortex imply this region is not affected by pathophysiological changes by the deletion of the gene. The analysis of SWD powers indicated that SWDs occurred predominantly in the medial prefrontal cortex in the PLCβ4 model. That the maximum latency from the initiator was much slower in the GHB model than the PLCβ4 model indicates fast propagated epileptogenic networks of the PLCβ4 model, whereas widespread networks are involved in SWD generation in the GHB model.
Comparison of topography
The pharmacological and genetic models used in this study showed markedly different spatiotemporal patterns in the propagation of SWDs in the brain. In the GHB model, the topographical analysis showed global and large-scale changes in power but the peak-to-peak amplitudes of spikes were relatively weaker. Interestingly, the spikes were confined primarily to the frontal cortex indicating increase of functional connectivity in this region. In contrast, topographical maps of the PLCβ4 model showed a strong signal in the prefrontal cortex, with a large spike amplitude in the parietal cortex, although the SWD was initiated in the somatosensory cortex. Increased functional connectivity in the prefrontal cortex is noticeable in the PLCβ4 model. In absence seizures in humans, functional connectivity increased between the frontal, parietal, and temporal lobes, whereas it decreased in the DMN, which may be attributed to the loss of consciousness [58]. In consciousness patients, the functional connectivity within the DMN has been known to be negatively correlated with the level of consciousness [59]. A consistent and reproducible observations in human absence seizure patients is a reduction in the activation of DMN structures and an enhancement in the activation of thalamus [1]. The blood oxygen level dependent signals in the medial and lateral prefrontal cortex, the posterior cingulate and the precuneus, the hub structures of DMN, were reported to be decreased during absence seizure [1, 13, 60]. Our data showed that the medial prefrontal cortex and parietal cortex belong to DMN experience frequent SWD in PLCβ4 model suggesting the model as consciousness patient model. But further analysis of functional connectivity between thalamus and cortex is required to translate this genetic model to human patients. Still, these finding indicates the possibility of developing therapeutics specifically to treat the consciousness patients. Relatively little activity of epileptic seizures in the parietal region and different spatiotemporal patterns in the GHB model may reflect that this pharmacological model represents conspicuously different SWD generation networks from the genetic models. The identification of an epileptogenic network is clinically important to improving the planning of surgery and targeted therapy to control abnormal activities in the relevant hubs and nodes of this network. We think comparisons between networks involved in absence generation in different animal models are valuable and relevant to clinical absence investigation in terms of identification of epileptogenic network and therapeutic target. Our data suggests that only parts of the consciousness networks seems to be involved in the GHB model; therefore, further verification is needed to establish that the GHB model is a clinically relevant absence epileptic model in future studies.
Supplementary Information
ACKNOWLEDGEMENTS
This research was supported by the National Research Foundation of Korea (NRF) grant funded by the Korean Government (MEST) (No.2012R1A5A2051385 to S Lee) and the National Research Foundation of Korea (NRF) grant funded by the Korea government (2017R1A2B3012659).
COMPETING INTERESTS STATEMENT
None.
AUTHOR CONTRIBUTIONS
SL wrote a manuscript, EH wrote a method and contributed editing, ML carried out experiments and JHC designed experiments, analyzed data and wrote a manuscript.
Figures
{kind=link}

{kind=link}
{kind=link}
References
- Carney PW, Jackson GD (2014) Insights into the mechanisms of absence seizure generation provided by EEG with functional MRI. Front Neurol 5 : 162.



- Lüttjohann A, van Luijtelaar G (2015) Dynamics of networks during absence seizure's on- and offset in rodents and man. Front Physiol 6 : 16.
- Williams D (1953) A study of thalamic and cortical rhythms in petit mal. Brain 76 : 50-69.
- Danober L, Deransart C, Depaulis A, Vergnes M, Marescaux C (1998) Pathophysiological mechanisms of genetic absence epilepsy in the rat. Prog Neurobiol 55 : 27-57.
- Meeren HK, Pijn JP, Van Luijtelaar EL, Coenen AM, Lopes da Silva FH (2002) Cortical focus drives widespread corticothalamic networks during spontaneous absence seizures in rats. J Neurosci 22 : 1480-1495.
- Manning JP, Richards DA, Leresche N, Crunelli V, Bowery NG (2004) Cortical-area specific block of genetically determined absence seizures by ethosuximide. Neuroscience 123 : 5-9.
- Polack PO, Guillemain I, Hu E, Deransart C, Depaulis A, Charpier S (2007) Deep layer somatosensory cortical neurons initiate spike-and-wave discharges in a genetic model of absence seizures. J Neurosci 27 : 6590-6599.
- Tan HO, Reid CA, Single FN, Davies PJ, Chiu C, Murphy S, Clarke AL, Dibbens L, Krestel H, Mulley JC, Jones MV, Seeburg PH, Sakmann B, Berkovic SF, Sprengel R, Petrou S (2007) Reduced cortical inhibition in a mouse model of familial childhood absence epilepsy. Proc Natl Acad Sci U S A 104 : 17536-17541.
- Stefan H (1997) Absence seizures. In: Epilepsy: a comprehensive textbook (Engel J, Pedley TA, eds), pp 590-597. Lippincott Williams & Wilkins, Philadelphia, PA.
- Holmes MD, Brown M, Tucker DM (2004) Are “generalized” seizures truly generalized? Evidence of localized mesial frontal and frontopolar discharges in absence. Epilepsia 45 : 1568-1579.
- Stefan H, Lopes da Silva FH (2013) Epileptic neuronal networks: methods of identification and clinical relevance. Front Neurol 4 : 8.
- Polack PO, Guillemain I, Hu E, Deransart C, Depaulis A, Charpier S (2007) Deep layer somatosensory cortical neurons initiate spike-and-wave discharges in a genetic model of absence seizures. J Neurosci 27 : 6590-6599.
- Sakurai K, Takeda Y, Tanaka N, Kurita T, Shiraishi H, Takeuchi F, Nakane S, Sueda K, Koyama T (2010) Generalized spikewave discharges involve a default mode network in patients with juvenile absence epilepsy: a MEG study. Epilepsy Res 89 : 176-184.
- Mazoyer B, Zago L, Mellet E, Bricogne S, Etard O, Houdé O, Crivello F, Joliot M, Petit L, Tzourio-Mazoyer N (2001) Cortical networks for working memory and executive functions sustain the conscious resting state in man. Brain Res Bull 54 : 287-298.
- Andreasen NC, O'Leary DS, Cizadlo T, Arndt S, Rezai K, Watkins GL, Ponto LL, Hichwa RD (1995) Remembering the past: two facets of episodic memory explored with positron emission tomography. Am J Psychiatry 152 : 1576-1585.
- McCormick DA, Bal T (1997) Sleep and arousal: thalamocortical mechanisms. Annu Rev Neurosci 20 : 185-215.
- Huntsman MM, Porcello DM, Homanics GE, DeLorey TM, Huguenard JR (1999) Reciprocal inhibitory connections and network synchrony in the mammalian thalamus. Science 283 : 541-543.
- Avanzini G, Panzica F, de Curtis M (2000) The role of the thalamus in vigilance and epileptogenic mechanisms. Clin Neurophysiol 111 Suppl 2 : S19-S26.
- Huguenard JR, McCormick DA (2007) Thalamic synchrony and dynamic regulation of global forebrain oscillations. Trends Neurosci 30 : 350-356.
- Beenhakker MP, Huguenard JR (2009) Neurons that fire together also conspire together: is normal sleep circuitry hijacked to generate epilepsy? Neuron 62 : 612-632.
- Steriade M (2005) Sleep, epilepsy and thalamic reticular inhibitory neurons. Trends Neurosci 28 : 317-324.
- Niedermeyer E (1965) Sleep electroencephalograms in petitmal. Arch Neurol 12 : 625-630.
- Halász P, Kelemen A (2009) New vistas and views in the concept of generalized epilepsies. Ideggyogy Sz 62 : 366-380.
- Pinault D, O'Brien TJ (2005) Cellular and network mechanisms of genetically-determined absence seizures. Thalamus Relat Syst 3 : 181-203.
- Pinault D, Slézia A, Acsády L (2006) Corticothalamic 5–9 Hz oscillations are more pro-epileptogenic than sleep spindles in rats. J Physiol 574 : 209-227.
- Meeren HK, Veening JG, Möderscheim TA, Coenen AM, van Luijtelaar G (2009) Thalamic lesions in a genetic rat model of absence epilepsy: dissociation between spike-wave discharges and sleep spindles. Exp Neurol 217 : 25-37.
- Depaulis A, Charpier S (2018) Pathophysiology of absence epilepsy: insights from genetic models. Neurosci Lett 667 : 53-65.
- Jarre G, Guillemain I, Deransart C, Depaulis A (2017) Chapter 32. Genetic models of absence epilepsy in rats and mice. In: Models of seizures and epilepsy. 2nd ed (Pitkänen A, Buckmaster PS, Galanopoulou AS, Moshé SL, eds), pp 455- 471. Academic Press, Oxford.
- Cortez MA, Kostopoulos GK, Snead OC 3rd (2016) Acute and chronic pharmacological models of generalized absence seizures. J Neurosci Methods 260 : 175-184.
- van Luijtelaar G (2017) Genetic models of absence epilepsy: new concepts and insights. In: Reference module in neuroscience and biobehavioral psychology (Stein J, ed). Elsevier, Amsterdam.
- Maheshwari A, Noebels JL (2014) Monogenic models of absence epilepsy: windows into the complex balance between inhibition and excitation in thalamocortical microcircuits. Prog Brain Res 213 : 223-252.
- Choi JH, Koch KP, Poppendieck W, Lee M, Shin HS (2010) High resolution electroencephalography in freely moving mice. J Neurophysiol 104 : 1825-1834.
- Cheong E, Zheng Y, Lee K, Lee J, Kim S, Sanati M, Lee S, Kim YS, Shin HS (2009) Deletion of phospholipase C β4 in thalamocortical relay nucleus leads to absence seizures. Proc Natl Acad Sci U S A 106 : 21912-21917.
- Absalom N, Eghorn LF, Villumsen IS, Karim N, Bay T, Olsen JV, Knudsen GM, Bräuner-Osborne H, Frølund B, Clausen RP, Chebib M, Wellendorph P (2012) α4βδ GABAA receptors are high-affinity targets for γ-hydroxybutyric acid (GHB). Proc Natl Acad Sci U S A 109 : 13404-13409.
- Cope DW, Di Giovanni G, Fyson SJ, Orbán G, Errington AC, Lőrincz ML, Gould TM, Carter DA, Crunelli V (2009) Enhanced tonic GABAA inhibition in typical absence epilepsy. Nat Med 15 : 1392-1398.
- Lee M, Kim D, Shin HS, Sung HG, Choi JH (2011) Highdensity EEG recordings of the freely moving mice using polyimide-based microelectrode. J Vis Exp (47):2562.
- Sutter R, Kaplan PW (2013) The neurophysiologic types of nonconvulsive status epilepticus: EEG patterns of different phenotypes. Epilepsia 54 Suppl 6:23-27.
- Kaplan PW (2006) The EEG of status epilepticus. J Clin Neurophysiol 23 : 221-229.
- Arcaro J, Ma J, Chu L, Kuo M, Mirsattari SM, Stan Leung L (2016) The hippocampus participates in a pharmacological rat model of absence seizures. Epilepsy Res 120 : 79-90.
- van Luijtelaar G, Sitnikova E (2006) Global and focal aspects of absence epilepsy: the contribution of genetic models. Neurosci Biobehav Rev 30 : 983-1003.
- Szaflarski JP, Kay B, Gotman J, Privitera MD, Holland SK (2013) The relationship between the localization of the generalized spike and wave discharge generators and the response to valproate. Epilepsia 54 : 471-480.
- Meeren H, van Luijtelaar G, Lopes da Silva F, Coenen A (2005) Evolving concepts on the pathophysiology of absence seizures: the cortical focus theory. Arch Neurol 62 : 371-376.
- Gloor P (1968) Generalized cortico-reticular epilepsies. Some considerations on the pathophysiology of generalized bilaterally synchronous spike and wave discharge. Epilepsia 9 : 249-263.
- Crunelli V, Leresche N (2002) Childhood absence epilepsy: genes, channels, neurons and networks. Nat Rev Neurosci 3 : 371-382.
- McCormick DA, Contreras D (2001) On the cellular and network bases of epileptic seizures. Annu Rev Physiol 63 : 815-846.
- Watanabe M, Nakamura M, Sato K, Kano M, Simon MI, Inoue Y (1998) Patterns of expression for the mRNA corresponding to the four isoforms of phospholipase Cβ in mouse brain. Eur J Neurosci 10 : 2016-2025.
- Cheong E, Lee S, Choi BJ, Sun M, Lee CJ, Shin HS (2008) Tuning thalamic firing modes via simultaneous modulation of Tand L-type Ca2+ channels controls pain sensory gating in the thalamus. J Neurosci 28 : 13331-13340.
- Pedroarena CM, Llinás R (2001) Interactions of synaptic and intrinsic electroresponsiveness determine corticothalamic activation dynamics. Thalamus Relat Syst 1 : 3-14.
- McCormick DA, von Krosigk M (1992) Corticothalamic activation modulates thalamic firing through glutamate “metabotropic” receptors. Proc Natl Acad Sci U S A 89 : 2774-2778.
- Pinault D, Leresche N, Charpier S, Deniau JM, Marescaux C, Vergnes M, Crunelli V (1998) Intracellular recordings in thalamic neurones during spontaneous spike and wave discharges in rats with absence epilepsy. J Physiol 509 : 449-456.
- Steriade M, Contreras D (1995) Relations between cortical and thalamic cellular events during transition from sleep patterns to paroxysmal activity. J Neurosci 15 : 623-642.
- Slaght SJ, Leresche N, Deniau JM, Crunelli V, Charpier S (2002) Activity of thalamic reticular neurons during spontaneous genetically determined spike and wave discharges. J Neurosci 22 : 2323-2334.
- Chen Y, Lu J, Pan H, Zhang Y, Wu H, Xu K, Liu X, Jiang Y, Bao X, Yao Z, Ding K, Lo WH, Qiang B, Chan P, Shen Y, Wu X (2003) Association between genetic variation of CACNA1H and childhood absence epilepsy. Ann Neurol 54 : 239-243.
- McCafferty C, David F, Venzi M, Lőrincz ML, Delicata F, Atherton Z, Recchia G, Orban G, Lambert RC, Di Giovanni G, Leresche N, Crunelli V (2018) Cortical drive and thalamic feed-forward inhibition control thalamic output synchrony during absence seizures. Nat Neurosci 21 : 744-756.
- Krook-Magnuson EI, Huntsman MM (2005) Excitability of cortical neurons depends upon a powerful tonic conductance in inhibitory networks. Thalamus Relat Syst 3 : 115-120.
- Venzi M, Di Giovanni G, Crunelli V (2015) A critical evaluation of the gamma-hydroxybutyrate (GHB) model of absence seizures. CNS Neurosci Ther 21 : 123-140.
- Pinard A, Seddik R, Bettler B (GABAB Receptors: Physiological Functions and Mechanisms of Diversity, in Advances in Pharmacology, Blackburn Thomas P., Editor. 2010, Academic Press. p. 231-255.
- Wu Q, Zhao CW, Long Z, Xiao B, Feng L (2018) Anatomy Based Networks and Topology Alteration in Seizure-Related Cognitive Outcomes. Front Neuroanat 12 : 25.
- Vanhaudenhuyse A, Noirhomme Q, Tshibanda LJ, Bruno MA, Boveroux P, Schnakers C, Soddu A, Perlbarg V, Ledoux D, Brichant JF, Moonen G, Maquet P, Greicius MD, Laureys S, Boly M (2010) Default network connectivity reflects the level of consciousness in non-communicative brain-damaged patients. Brain 133 : 161-171.
- Danielson NB, Guo JN, Blumenfeld H (2011) The default mode network and altered consciousness in epilepsy. Behav Neurol 24 : 55-65.