Articles

Article Tools
 View Full Text View Full Text |
 Abstract Abstract |
 Article as PDF Article as PDF |
 Print this Article Print this Article |
 Pubmed Pubmed |
 PMC PMC |
 PubReader PubReader |
 Export to Citation Export to Citation |
 Email Alerts Email Alerts |
 Open Access Open Access |
Share this article on :



Stats or Metrics
Article
Case Report
Exp Neurobiol 2016; 25(6): 347-350
Published online December 31, 2016
https://doi.org/10.5607/en.2016.25.6.347
© The Korean Society for Brain and Neural Sciences
Rapid Progression of Sporadic ALS in a Patient Carrying SOD1 p.Gly13Arg Mutation
Myung-Jin Kim1, Jae-Han Bae1, Jeong-Min Kim1, Hye Ryoun Kim2, Byung-Nam Yoon3, Jung-Joon Sung4 and Suk-Won Ahn1*
1Department of Neurology, Chung-Ang University Hospital, Chung-Ang University College of Medicine, 2Department of Laboratory Medicine, Chung-Ang University Hospital, Chung-Ang University College of Medicine, Seoul 06973, 3Department of Neurology, Inha University Hospital, Inha University College of Medicine, Incheon 22332, 4Department of Neurology, Seoul National University Hospital, Seoul National University College of Medicine, Seoul 03080, Korea
Correspondence to: *To whom correspondence should be addressed.
TEL: 82-2-6299-3153, FAX: 82-2-6299-3153
e-mail: icandr@hanmail.net
Abstract
Amyotrophic lateral sclerosis (ALS), the most common adult onset motor neuron disease, is pathologically characterized by progressive loss of the upper and lower motor neurons. Mutations in the Cu/Zn superoxide dismutase gene (SOD1) account for about 20% of familial ALS cases and a small percentage of sporadic ALS (SALS) cases, and have revealed a validated genotype-phenotype correlation. Herein, we report a p.Gly13Arg mutation in SOD1 exon 1 in a patient with SALS who presented with a rapidly progressive course, predominantly affecting the lower motor neurons. A 48-year-old man presented with progressive weakness and muscle atrophy of the left upper and lower limbs, followed by muscle fasciculation and cramping. The clinical features of the patient were clearly suggestive of ALS, and implied a sporadic form with rapid progression, predominantly affecting the lower motor neurons. Sequencing of the SOD1 gene by PCR revealed a missense mutation of G to C (c.37G>C) in exon 1, and amino acid substitution of glycine by arginine (p.Gly13Arg). This is the first case identifying the p.Gly13Arg mutation of SOD1 in the Korean population, and clinical assessments of this patient revealed a different phenotype compared with other cases.
Keywords: Amyotrophic lateral sclerosis, ALS, Cu/Zn superoxide dismutase 1, SOD1, Exon 1
INTRODUCTION
Amyotrophic lateral sclerosis (ALS), the most common adult onset motor neuron disease, is pathologically characterized by progressive loss of the upper and lower motor neurons in the brainstem motor nuclei and the anterior horn of the spinal cord [1,2]. This pattern of neurodegeneration produces progressive weakness, muscular wasting, and spasticity. The disease starts segmentally before it spreads and causes death from respiratory failure or infection a few years after its onset [3,4]. However, the modalities and prognosis of disease progression are clinically diverse, including involvement of bulbar muscle and presence of gene mutations [5,6,7].
Most patients with ALS present with the sporadic form (SALS) of uncertain or degenerative etiology, while approximately 5%~10% of ALS cases are classified as familial (FALS) [5,6,7,8]. To date, a number of genetic loci and disease-causing mutations in several genes, including the Cu/Zn superoxide dismutase gene (SOD1), C9orf72, TARDBP, FUS, OPTN, VCP, UBQLN2, and PFN1, have been reported to be associated with familial and sporadic ALS cases [5,6,7,8,9]. Among these, mutations of SOD1 account for about 15%~20% of all FALS and 2%~4% of all SALS cases, and have revealed a validated genotype-phenotype correlation [8,9,10].
Herein, we report a p.Gly13Arg mutation in SOD1 exon 1 in a patient with SALS who presented with a rapidly progressive course, predominantly affecting the lower motor neurons.
A 48-year-old man presented with progressive weakness and muscle atrophy of the left upper and lower limbs, followed by muscle fasciculation and cramping over the course of 5 months. There was no familial history of neuromuscular disease. Both the patient’s parents were still alive without symptoms of muscular weakness or atrophy. Upon e valuation, t he Medical R esource Council (MRC) scale revealed 4/5 in the left upper and lower limbs, and prominent muscular atrophy was observed in the left limbs compared to the right limbs. However, other neurological evaluations involving cognition, cranial nerve function, sensory system, speech, and swallowing revealed no abnormality. In addition, the deep tendon reflexes (DTR) of 4-limbs revealed normal or mildly decreased responses, and there were no pathological reflexes such as Babinski and Hoffman's signs.
Laboratory investigations revealed a normal hemogram, serum electrolytes, liver, thyroid and renal functions with the exception of a mildly elevated serum creatine kinase (CK) level (352 IU/L, normal range; 0-190). The results of tumor screening tests involving alpha-fetoprotein, carcinoembryonic antigen, and cancer antigen (CA)-19-9 were normal. Evaluations for vasculitis involving rheumatoid factor, anti-dsDNA antibody, lupus anticoagulant, anticardiolipin antibody, antineutrophil cytoplasmic antibody, anti-SSA and SSB (Sjogren syndrome A and B) antibody, anti-jo-1 antibody and anti-GM1 antibody were within the normal range or negative. Brain magnetic resonance imaging and angiography (MRI and MRA), spinal MRI, Jolly test, and routine nerve conduction study (NCS) were unremarkable.
Electromyography (EMG) of the vastus lateralis, tibialis anterior, peroneus longus, biceps brachii, triceps brachii, first dorsal interosseous, and paraspinal muscles showed large motor unit potentials and denervation potentials of fibrillation and positive sharp waves which were indicative of ALS or other motor neuron diseases [2,4,11].
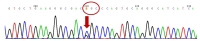
After obtaining informed consent for genetic examination, we tested the SOD1 gene in genomic DNA extracted from peripheral lymphocytes, and a missense mutation of G to C (c.37G>C) in exon 1, and amino acid substitution of arginine for glycine(p.Gly13Arg) was identified by using the sequencing of the SOD1 gene by PCR (Fig. 1).
Clinical findings of our patient were compatible with motor neuron disease (specifically ALS), predominantly affecting the lower motor neurons. The ALS functional rating score (ALSFRS) was 42/48 at 5 months after clinical onset.
Three months after diagnosis, the patient’s symptoms progressed rapidly involving worsening weakness with atrophy, dysarthria and dysphagia. So the patient became wheel-chair bound, and ALSFRS was decreased to 32/48 at 3 months after the initial diagnosis of ALS.
DISCUSSION
To date, more than 180 different mutations in the SOD1 gene have been observed in approximately 390 ALS patients (female; 45%, male; 54%), and the mean age of onset was 48 years for patients with a SOD1 mutation (http://alsod.iop.kcl.ac.uk/).
Among these SOD1 mutations, percentage distribution of FALS and SALS was 86 % and 13 %, respectively, and the proportions of limb and bulbar onset were 89% and 5%, respectively (http://alsod.iop.kcl.ac.uk/).
In this report, we describe an ALS patient with the p.Gly13Arg mutation in SOD1 exon 1, who presented with a sporadic form of the disease and a rapid progressive course over 8 months, with attention to clinical manifestations and genetic evaluation.
A missense mutation of G to C (c.37G>C) in exon 1 and amino acid substitution of arginine for glycine at position 13 (p.Gly13Arg) had been identified in an Italian patient with FALS previously [12,13].
To our knowledge, our patient is the second ALS case with the p.Gly13Arg mutation since that report; furthermore, this is the first report identifying this mutation in the Korean population [8,9]. Interestingly, the clinical implications of this case revealed a different phenotype of the sporadic form of ALS and clinical course, including onset age and rapid progression as compared with the Italian case [12,13,14].
In the Italian case, a male patient developed weakness of the left lower limb at age 63. At age 67, neurological examination showed mild weakness and wasting of the distal muscles in the lower limbs with muscle fasciculation. The ankle jerks were absent and plantar responses were normal with no signs of bulbar involvement. A similarly slow progression was reported in the patient’s father, who also presented with primary involvement of the lower limb muscles, followed by bulbar symptoms found at 4 years after disease onset [12,13].
Our patient showed predominantly lower motor neuron signs without apparent upper motor neuron signs which indicate the similarities with the Italian case. Therefore, this clinical feature indicates a possible clinic-genetic phenotype of ALS patients with a p.Gly13Arg mutation of SOD1. However, our patient showed a younger onset age (age 48 versus age 62), and had no apparent familial history of ALS. The patient’s parents were still alive without motor neuron signs. In addition to the different age of onset and familial history, our patient presented a differentiated clinical feature that was rapid progressive course with predominant involvement of lower motor neurons, while the Italian patient had a slower progressive course with predominantly lower motor neuron signs.
In conclusion, this case of p.Gly13Arg SOD1 mutation in SALS differs clinically from previous report of FALS, and expands the variations of ALS phenotypes associated with SOD1 gene mutations. To our knowledge, this is the first report identifying the p.Gly13Arg mutation of SOD1 in the Korean population. Further clinical assessments on the patient’s disease course and genetic analysis of related subjects will be necessary to validate the clinical characteristics of ALS with a p.Gly13Arg SOD1 mutation in the Korean population.
Figures
{kind=link}
References
- Kim C, Lee HC, Sung JJ. Amyotrophic lateral sclerosis - cell based therapy and novel therapeutic development. Exp Neurobiol 2014;23:207-214.

- Ahn SW, Kim SH, Oh DH, Kim SM, Park KS, Hong YH, Kwon OS, Sung JJ, Lee KW. Motor unit number estimation in evaluating disease progression in patients with amyotrophic lateral sclerosis. J Korean Med Sci 2010;25:1359-1363.
- Kim DG, Hong YH, Shin JY, Lee KW, Park KS, Seong SY, Sung JJ. Pattern of respiratory deterioration in sporadic amyotrophic lateral sclerosis according to onset lesion by using respiratory function tests. Exp Neurobiol 2015;24:351-357.
- Ahn SW, Kim SH, Kim JE, Kim SM, Kim SH, Park KS, Sung JJ, Lee KW, Hong YH. Reproducibility of the motor unit number index (MUNIX) in normal controls and amyotrophic lateral sclerosis patients. Muscle Nerve 2010;42:808-813.
- Rosen DR, Siddique T, Patterson D, Figlewicz DA, Sapp P, Hentati A, Donaldson D, Goto J, O’Regan JP, Deng HX, Rahmani Z, Krizus A, McKenna-Yasek D, Cayabyab A, Gaston SM, Berger R, Tanzi RE, Halperin JJ, Herzfeldt B, Van den Bergh R, Hung WY, Bird T, Deng G, Mulder DW, Smyth C, Laing NG, Soriano E, Pericak-Vance MA, Haines J, Rouleau GA, Gusella JS, Horvitz HR, Brown RH. Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature 1993;362:59-62.
- Chiò A, Traynor BJ, Lombardo F, Fimognari M, Calvo A, Ghiglione P, Mutani R, Restagno G. Prevalence of SOD1 mutations in the Italian ALS population. Neurology 2008;70:533-537.
- Kim NH, Kim HJ, Kim M, Lee KW. A novel SOD1 gene mutation in a Korean family with amyotrophic lateral sclerosis. J Neurol Sci 2003;206:65-69.
- Kim HJ, Oh KW, Kwon MJ, Oh SI, Park JS, Kim YE, Choi BO, Lee S, Ki CS, Kim SH. Identification of mutations in Korean patients with amyotrophic lateral sclerosis using multigene panel testing. Neurobiol Aging 2016;37:209.e9-209.e16.
- Kwon MJ, Baek W, Ki CS, Kim HY, Koh SH, Kim JW, Kim SH. Screening of the SOD1, FUS, TARDBP, ANG, and OPTN mutations in Korean patients with familial and sporadic ALS. Neurobiol Aging 2012;33:1017.e17-1017.e23.
- Takazawa T, Ikeda K, Hirayama T, Kawabe K, Nakamura Y, Ito H, Kano O, Yoshii Y, Tanaka F, Sobue G, Iwasaki Y. Familial amyotrophic lateral sclerosis with a novel G85S mutation of superoxide dismutase 1 gene: clinical features of lower motor neuron disease. Intern Med 2010;49:183-186.
- Yoon BN, Choi SH, Rha JH, Kang SY, Lee KW, Sung JJ. Comparison between flail arm syndrome and upper limb onset amyotrophic lateral sclerosis: clinical features and electromyographic findings. Exp Neurobiol 2014;23:253-257.
- Penco S, Schenone A, Bordo D, Bolognesi M, Abbruzzese M, Bugiani O, Ajmar F, Garrè C. A SOD1 gene mutation in a patient with slowly progressing familial ALS. Neurology 1999;53:404-406.
- Gellera C, Castellotti B, Riggio MC, Silani V, Morandi L, Testa D, Casali C, Taroni F, Di Donato S, Zeviani M, Mariotti C. Superoxide dismutase gene mutations in Italian patients with familial and sporadic amyotrophic lateral sclerosis: identification of three novel missense mutations. Neuromuscul Disord 2001;11:404-410.
- Nakamura A, Kuru S, Hineno A, Kobayashi C, Kinoshita T, Miyazaki D, Ikeda S. Slowly progressing lower motor neuron disease caused by a novel duplication mutation in exon 1 of the SOD1 gene. Neurobiol Aging 2014;35:2420.e7-2420.e12.