Articles

Article Tools
 View Full Text View Full Text |
 Abstract Abstract |
 Article as PDF Article as PDF |
 Print this Article Print this Article |
 Pubmed Pubmed |
 PMC PMC |
 PubReader PubReader |
 Export to Citation Export to Citation |
 Email Alerts Email Alerts |
 Open Access Open Access |
Share this article on :



Stats or Metrics
Article
Original Article
Exp Neurobiol 2018; 27(6): 550-563
Published online December 12, 2018
https://doi.org/10.5607/en.2018.27.6.550
© The Korean Society for Brain and Neural Sciences
A De Novo RAPGEF2 Variant Identified in a Sporadic Amyotrophic Lateral Sclerosis Patient Impairs Microtubule Stability and Axonal Mitochondria Distribution
Keunjung Heo1,†, Su Min Lim2,†, Minyeop Nahm2,†, Young-Eun Kim3, Ki-Wook Oh2, Hwan Tae Park4, Chang-Seok Ki5*, Seung Hyun Kim2*, and Seungbok Lee1*
1Department of Brain and Cognitive Sciences and Dental Research Institute, Seoul National University, Seoul 08826, Korea.
2Department of Neurology, College of Medicine, Hanyang University, Seoul 04763, Korea.
3Department of Laboratory Medicine, College of Medicine, Hanyang University, Seoul 04763, Korea.
4Department of Molecular Neuroscience, College of Medicine, Dong-A University, Busan 49201, Korea.
5Green Cross Genome Corporation, Yongin 16924, Korea.
Correspondence to: *To whom correspondence should be addressed.
Chang-Seok Ki, TEL: 82-31-260-0601, FAX: 82-31-260-9087, e-mail: changski.md@gmail.com
Seung Hyun Kim, TEL: 82-2-2290-8371, FAX: 82-2-2296-8370, e-mail: kimsh1@hanyang.ac.kr
Seungbok Lee, TEL: 82-2-880-2330, FAX: 82-2-762-2583, e-mail: seunglee@snu.ac.kr
†
These authors contributed equally to this work.
Abstract
Amyotrophic lateral sclerosis (ALS) is a fatal neurodegenerative disease that is frequently linked to microtubule abnormalities and mitochondrial trafficking defects. Whole exome sequencing (WES) of patient-parent trios has proven to be an efficient strategy for identifying rare
Graphical Abstract
Keywords: Amyotrophic lateral sclerosis, Whole exome sequencing,
INTRODUCTION
Amyotrophic lateral sclerosis (ALS) is a progressive neurodegenerative disease that affects both upper and lower motor neurons, leading to muscle weakness and atrophy followed by paralysis [1,2]. ALS is usually fatal due to respiratory failure within 5 years after symptom onset and represents the most common form of adult-onset motor neuron diseases with an incidence of 2 per 100,000. Approximately 10% of cases show familial inheritance, while the remaining majority of cases occur sporadically. Over the last two decades, substantial progress has been made in understanding of the genetic landscape of familial ALS (fALS). To date, two-thirds of fALS are associated with mutations in any of more than 25 genes [3,4,5], encoding proteins involved in protein homeostasis, RNA metabolism, vesicular trafficking, and cytoskeletal organization. Despite this progress in identifying fALS-associated genes, the genetic etiology of sporadic ALS (sALS) remains largely unknown [3].
Impaired mitochondrial trafficking in motor neurons is a well-established phenomenon in ALS pathophysiology [6]. Electron microscopic studies of post-mortem ALS cases demonstrated remarkable accumulation of mitochondria in the somata and proximal axons of motor axons in the spinal cord [7]. Consistently, abnormal clustering of mitochondria in proximal axons of motor neurons was also observed in transgenic mice and rats expressing the ALS mutant SOD1-G93A [8,9]. Similar defects in mitochondrial distribution were also observed in motor neurons from transgenic mice expressing ALS-associated TDP-43 mutants [10,11]. However, the underlying mechanisms of mitochondrial trafficking defects in ALS remain to be fully understood.
Whole exome sequencing (WES) has significantly contributed to our knowledge of ALS genetics. First, WES studies on ALS families identified novel pathogenic variants in known fALS genes, including
In this study, we employed a trio-WES approach to identify pathogenic variants in a sALS patient and detected a
MATERIALS AND METHODS
The 28-year-old female patient and her healthy parents provided written informed consent as approved by the Institutional Review Boards of Hanyang University Hospital (Seoul, Korea). Genomic DNA was isolated from peripheral blood leukocytes using the Wizard Genomic DNA Purification kit (Promega, Madison, WI, USA). The exomes of subjects were captured using the Agilent SureSelect all Exon 50Mb kit (Agilent, Santa Clara, CA, USA) and sequenced on an Illumina NextSeq500 machine (paired-end and 100-bp reads) (Illumina, San Diego, CA, USA). Reads were mapped to a custom GRCh37/hg19 build using the Burrows-Wheeler Aligner (BWA). Annotation was performed using an in-house custom-made script. We selected rare variants with allele frequency less than 0.01 identified in the NHLBI Exome Sequencing Project (http://evs.gs.washington.edu/EVS/), the 1000 Genomes Project (http://www.1000genomes.org/), and gnomAD (http://gnomad.broadinstitute.org/). All amino acid-altering
Full-length cDNAs for
Primary human fibroblasts were established from punch biopsies on the forearm skin of the patient and a 43-year-old male control as described previously [23] and maintained in Dulbecco's modified Eagle's medium (DMEM) containing 20% heat-inactivated (30 min, 55℃) fetal bovine serum (FBS), 1% non-essential amino acids, and antibiotics. Passage-matched control and patient fibroblasts (prior to passage 10) were used in each experiment. For inhibition of HDAC6, human skin fibroblasts were treated with 1 µM tubastatin A (Sigma-Aldrich, St. Louis, MI, USA) overnight at 37℃. Human HeLa cells were maintained in DMEM supplemented with 10% heat-inactivated FBS and transfected using FuGENE HD transfection reagent (Promega, Madison, WI, USA).
Flies were maintained at 25℃ on standard food. Transgenic
Cultured cells were fixed with 4% formaldehyde in PBS for 20 min at room temperature, permeabilized with 0.2% Triton X-100 in PBS for 10 min, and blocked with 1% BSA in PBS for 1 h. Samples were then incubated with primary antibodies for 1 h and sequentially incubated with fluorescently labeled secondary antibodies for 30 min at room temperature. Wandering third-instar
Fluorescent images were acquired with an LSM 700 laser-scanning confocal microscope using a C Apo 40x W or Plan Apo 63x 1.4 NA objective (Carl Zeiss, Jena, Germany). The length of Mito-RFP-labeled mitochondria in fibroblasts was determined using ImageJ. For quantification of the number and area of mitochondria in
A climbing test was used to assess the locomotor function of adult flies as previously described [24]. Briefly, 45 adult flies aged for 20 days were transferred into a glass graduated cylinder. Following 5-min acclimation, flies were gently tapped to the bottom and the distance climbed by individual flies in a 30 s period was measured.
Primary human skin fibroblasts were homogenized in ice-cold lysis buffer (50 mM Tris-HCl, pH 7.4, 150 mM NaCl, 1% Triton X-100, and protease inhibitors) and subjected to western blotting as previously described [23]. For some experiments, we separated the mitochondrial and cytosolic fractions from fibroblast lysates using the BioVision Mitochondria/Cytosol Fractionation kit (BioVision, Milpitas, CA, USA). The following primary antibodies were used: anti-acetylated α-tubulin (1:1000; Sigma-Aldrich), anti-tyrosinated α-tubulin (1:1000; Millipore), anti-α-tubulin (1:1000; Sigma-Aldrich), anti-BAX (1:1000; BD, Franklin Lakes, NJ, USA), and anti-GAPDH (1:1000; Santa Cruz Biotechnology, Dallas, TX, USA).
The mitochondrial membrane potential was assessed in live primary fibroblasts using 5,5′,6,6′-tetrachloro-1,1′,3,3′-tetraethylbenzimidazolylcarbocyanine iodide (JC-1; Sigma-Aldrich) as described [26]. Briefly, fibroblasts were washed and incubated with 5 µg/ml JC-1 dye for 20 min at 37℃. The cells were then rinsed with culture medium, and their images were obtained using the Applied Precision DeltaVision fluorescence microscopy system (GE Healthcare, Chicago, IL, USA). JC-1 accumulates as red fluorescent aggregates within polarized mitochondria but does as green fluorescent monomers within less polarized mitochondria.
Primary human skin fibroblasts were fixed in PBS containing 4% paraformaldehyde and 2.5% glutaraldehyde for 24 h and rinsed in PBS. The samples were then subjected to 70-nm sectioning after gradual dehydration in ethanol solutions and propylene oxide (Acros Organics, Morris Plains, NJ, USA), and stained with epoxy resins using standard procedures. Images were acquired with a Hitachi electron microscope (Hitachi, Tokyo, Japan) equipped with a ES500W digital camera (GATAN, Pleasanton, CA, USA).
Comparisons were made by one-way ANOVA analysis with a post-hoc Turkey test. Data are presented as mean±SEM.
RESULTS
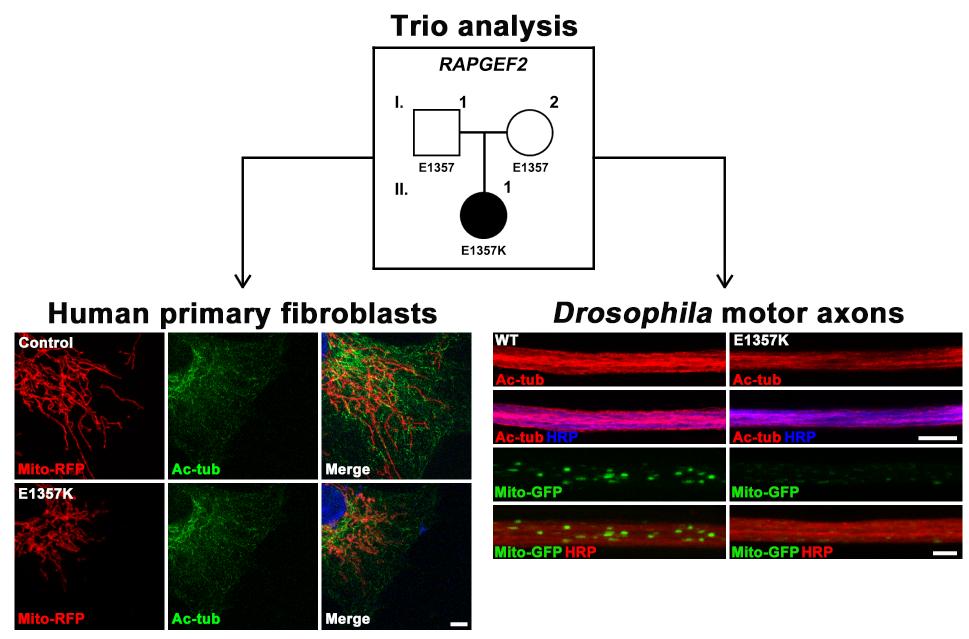
While performing WES on sALS trios, we identified a 28-year-old female patient carrying a
The
Disruptions in microtubule network assembly have been proposed as a critical component of ALS pathogenesis [22]. We therefore asked whether the E1357K variant of RAPGEF2 affects microtubule dynamics or organization. To address this, we visualized microtubule networks in control- and patient-derived skin fibroblasts using antibodies against α-tubulin (detecting both free α-tubulin and microtubules), acetylated α-tubulin (detecting long-lived, stable microtubules [31]), and tyrosinated α-tubulin (detecting both free tubulin and newly formed microtubules [32]). In control cells, all of these tubulin antibodies revealed the typical microtubule pattern of an aster-like distribution extending toward the cell periphery (Fig. 2A). The levels and distribution of α-tubulin and tyrosinated α-tubulin remained unchanged in patient fibroblasts (Fig. 2A). In sharp contrast, anti-acetylated α-tubulin signals were weaker in patient fibroblasts than in control fibroblasts (Fig. 2A). In addition, acetylated α-tubulin networks were restricted only in the perinuclear region. We confirmed the selective reduction of acetylated α-tubulin in patient cells by western blotting (Fig. 2B~D). These results suggest that the E1357K variant of RAPGEF2 affects the stability of the microtubule network.
To corroborate the above conclusion, we analyzed the effect of the RAPGEF2-E1357K variant on the level of α-tubulin acetylation in HeLa cells. Levels of anti-acetylated α-tubulin signal were significantly decreased in RAPGEF2-E1357K-transfected cells compared with untransfected control cells (Fig. 2E), confirming a deleterious effect of the RAPGEF2 variant on microtubule stability.
Since appropriate mitochondrial distribution critically depends on microtubule-based transport, we investigated whether alteration of the microtubule network in patient-derived fibroblasts is paralleled with abnormalities in mitochondrial morphology and distribution. Control and patient fibroblasts were transfected with a mitochondrial matrix-localized RFP (Mito-RFP) reporter construct. In control cells, Mito-RFP signals largely appeared as tubular networks extending throughout the cytoplasm (Fig. 3A). In contrast, the Mito-RFP-labeled mitochondria networks in patient-derived cells were fragmented and more restricted around the perinuclear area (Fig. 3A). To quantify mitochondrial fragmentation, we measured mitochondrial length. Patient fibroblasts showed a reduction of about 88% in average mitochondrial length compared with control cells (Fig. 3B). However, the size of cells was comparable between both genotypes (Fig. 3C).
Next, we investigated whether the decrease in the stability of microtubules is a causative mechanism of abnormal mitochondrial distribution in patient-derived fibroblasts. Acetylation of lysine 40 in α-tubulin, an indication of microtubule stabilization, is increased by inhibiting the catalytic activity of histone deacetylase 6 (HDAC6) [33], which is the major deacetylase of α-tubulin [34]. We examined the effect of an HDAC6-selective inhibitor (tubastatin A) on mitochondrial distribution in patient fibroblasts. Treatment of fibroblasts with 1 µM tubastatin A restored abnormalities in acetylated α-tubulin distribution and mitochondrial distribution and length (Fig. 3A~C). These results suggest that impaired microtubule stability is a causative mechanism accounting for abnormal mitochondrial distribution in patient-derived fibroblasts expressing the RAPGEF2-E1357K variant.
To validate the effects of the RAPGEF2-E1357K mutant on microtubule maintenance and mitochondrial distribution in motor axons, we generated
We have recently shown that loss of the
To test if expression of the RAPGEF2-E1357K mutant induces motor dysfunction, we performed climbing assays on adult flies at 20 days of age. Compared with transgenic controls (
To investigate the effects of the RAPGEF2-E1357K variant on mitochondrial structures, we performed transmission electron microscopy (EM) for control and patient fibroblasts. Control mitochondria showed a typical crista structure with electron-dense deposits in the matrix (Fig. 6A). In contrast, patient fibroblasts displayed swollen and vacuolated mitochondria without lamella cristae and electron-dense deposits (Fig. 6A).
To investigate whether the ultrastructural mitochondrial abnormalities in patient fibroblasts are paralleled with mitochondrial dysfunction, we assessed mitochondrial membrane potential using JC-1, a cationic lipophilic dye. The JC-1 dye accumulates as red-fluorescent J-aggregates within mitochondria at high membrane potentials (energized mitochondria), while it accumulates as green fluorescent monomers within mitochondria at low membrane potentials (deenergized mitochondria) [36]. In live control fibroblasts loaded with JC-1, we observed strong red fluorescent signals but not green fluorescent signals (Fig. 6B), suggesting the majority of mitochondria are functional. In contrast, both red and green fluorescent signals were prominent in JC-1-loaded patient fibroblasts (Fig. 6B). These results suggest that mitochondria activity is lower in patient fibroblasts than in control fibroblasts.
Mitochondrial dysfunction is intimately linked to apoptotic cell death, which involves the mitochondrial recruitment of the proapoptotic regulator BAX from the cytosol [37]. We therefore examined whether mitochondrial dysfunction in patient-derived skin fibroblasts is paralleled with abnormal accumulation of BAX on the mitochondria. To this end, we transfected control and patient fibroblasts with a GFP-BAX construct and stained them with anti-GFP and anti-mitochondria. In control fibroblasts, BAX showed a diffuse cytoplasmic distribution with a minimal overlap with mitochondria (Fig. 7A). Notably, we observed a prominent overlap between BAX signals and mitochondria in patient fibroblasts (Fig. 7A). As an additional approach, we separated cytosolic and mitochondrial fractions from lysates of control and patient fibroblasts and performed Western blot analysis of these fractions using anti-BAX. This experiment demonstrated that BAX was more recruited to the mitochondria in patient fibroblasts than in control fibroblasts (Fig. 7B and C). In a control experiment, we confirmed that total BAX levels were comparable between control and patient derived fibroblasts (data not shown). Finally, we found that the abnormal accumulation of BAX at the mitochondria was rescued by inhibiting HDAC6 with 1 µM tubastatin A (Fig. 7A~C). The same dose of tubastatin A had no effect on the BAX distribution in control-derived fibroblasts (Fig. 7A~C).
DISCUSSION
In this study, we report the identification of a
Microtubules are dynamic polymers that undergo polymerization and depolymerization of α- and β-tubulin heterodimer. While developing neurons keep microtubules in a highly dynamic state during process outgrowth, mature neurons progressively increase microtubule stability to maintain many aspects of cellular functions [39,40]. This change in microtubule stability is associated with chemical modifications of microtubules including acetylation [39]. Microtubule acetylation and deacetylation mainly occur at the conserved lysine 40 residue of α-tubulin by α-tubulin acetyltransferase 1 (αTAT1) and histone deacetylase 6 (HDAC6), respectively [34,41]. Interestingly, several studies have demonstrated HDAC6-mediated modulation of ALS pathogenesis. For example, removal of the
Consistent with our previous findings [24], knockdown of a
In conclusion, we identified a
Figures
{kind=link}

{kind=link}

{kind=link}

{kind=link}

{kind=link}

{kind=link}

{kind=link}
References
- Robberecht W, Philips T. The changing scene of amyotrophic lateral sclerosis. Nat Rev Neurosci 2013;14:248-264.

- Taylor JP, Brown RH, Cleveland DW. Decoding ALS: from genes to mechanism. Nature 2016;539:197-206.
- Renton AE, Chiò A, Traynor BJ. State of play in amyotrophic lateral sclerosis genetics. Nat Neurosci 2014;17:17-23.
- Müller K, Brenner D, Weydt P, Meyer T, Grehl T, Petri S, Grosskreutz J, Schuster J, Volk AE, Borck G, Kubisch C, Klopstock T, Zeller D, Jablonka S, Sendtner M, Klebe S, Knehr A, Günther K, Weis J, Claeys KG, Schrank B, Sperfeld AD, Hübers A, Otto M, Dorst J, Meitinger T, Strom TM, Andersen PM, Ludolph AC, Weishaupt JH, German ALS network MND-NET. Comprehensive analysis of the mutation spectrum in 301 German ALS families. J Neurol Neurosurg Psychiatry 2018;89:817-827.
- Leblond CS, Kaneb HM, Dion PA, Rouleau GA. Dissection of genetic factors associated with amyotrophic lateral sclerosis. Exp Neurol 2014;262:91-101.
- De Vos KJ, Hafezparast M. Neurobiology of axonal transport defects in motor neuron diseases: opportunities for translational research?. Neurobiol Dis 2017;105:283-299.
- Sasaki S, Iwata M. Mitochondrial alterations in the spinal cord of patients with sporadic amyotrophic lateral sclerosis. J Neuropathol Exp Neurol 2007;66:10-16.
- De Vos KJ, Chapman AL, Tennant ME, Manser C, Tudor EL, Lau KF, Brownlees J, Ackerley S, Shaw PJ, McLoughlin DM, Shaw CE, Leigh PN, Miller CC, Grierson AJ. Familial amyotrophic lateral sclerosis-linked SOD1 mutants perturb fast axonal transport to reduce axonal mitochondria content. Hum Mol Genet 2007;16:2720-2728.
- Sotelo-Silveira JR, Lepanto P, Elizondo V, Horjales S, Palacios F, Martinez-Palma L, Marin M, Beckman JS, Barbeito L. Axonal mitochondrial clusters containing mutant SOD1 in transgenic models of ALS. Antioxid Redox Signal 2009;11:1535-1545.
- Magrané J, Sahawneh MA, Przedborski S, Estévez AG, Manfredi G. Mitochondrial dynamics and bioenergetic dysfunction is associated with synaptic alterations in mutant SOD1 motor neurons. J Neurosci 2012;32:229-242.
- Wang W, Li L, Lin WL, Dickson DW, Petrucelli L, Zhang T, Wang X. The ALS disease-associated mutant TDP-43 impairs mitochondrial dynamics and function in motor neurons. Hum Mol Genet 2013;22:4706-4719.
- Daoud H, Zhou S, Noreau A, Sabbagh M, Belzil V, Dionne-Laporte A, Tranchant C, Dion P, Rouleau GA. Exome sequencing reveals SPG11 mutations causing juvenile ALS. Neurobiol Aging 2012;33:839.e5-839.e9.
- Klein CJ, Wu Y, Duan X, Middha S, Dawson BD, Kocher JP, Dyck PJ. Novel SOD1 mutation discovered in atypical ALS by whole exome sequencing. J Neurol Neurosurg Psychiatry 2013;84:943-944.
- Williams KL, Warraich ST, Yang S, Solski JA, Fernando R, Rouleau GA, Nicholson GA, Blair IP. UBQLN2/ubiquilin 2 mutation and pathology in familial amyotrophic lateral sclerosis. Neurobiol Aging 2012;33:2527.e3-2527.e10.
- Johnson JO, Mandrioli J, Benatar M, Abramzon Y, Van Deerlin VM, Trojanowski JQ, Gibbs JR, Brunetti M, Gronka S, Wuu J, Ding J, McCluskey L, Martinez-Lage M, Falcone D, Hernandez DG, Arepalli S, Chong S, Schymick JC, Rothstein J, Landi F, Wang YD, Calvo A, Mora G, Sabatelli M, Monsurrò MR, Battistini S, Salvi F, Spataro R, Sola P, Borghero G, Galassi G, Scholz SW, Taylor JP, Restagno G, Chiò A, Traynor BJ, ITALSGEN Consortium. Exome sequencing reveals VCP mutations as a cause of familial ALS. Neuron 2010;68:857-864.
- Kim HJ, Kim NC, Wang YD, Scarborough EA, Moore J, Diaz Z, MacLea KS, Freibaum B, Li S, Molliex A, Kanagaraj AP, Carter R, Boylan KB, Wojtas AM, Rademakers R, Pinkus JL, Greenberg SA, Trojanowski JQ, Traynor BJ, Smith BN, Topp S, Gkazi AS, Miller J, Shaw CE, Kottlors M, Kirschner J, Pestronk A, Li YR, Ford AF, Gitler AD, Benatar M, King OD, Kimonis VE, Ross ED, Weihl CC, Shorter J, Taylor JP. Mutations in prion-like domains in hnRNPA2B1 and hnRNPA1 cause multisystem proteinopathy and ALS. Nature 2013;495:467-473.
- Wu CH, Fallini C, Ticozzi N, Keagle PJ, Sapp PC, Piotrowska K, Lowe P, Koppers M, McKenna-Yasek D, Baron DM, Kost JE, Gonzalez-Perez P, Fox AD, Adams J, Taroni F, Tiloca C, Leclerc AL, Chafe SC, Mangroo D, Moore MJ, Zitzewitz JA, Xu ZS, van den Berg LH, Glass JD, Siciliano G, Cirulli ET, Goldstein DB, Salachas F, Meininger V, Rossoll W, Ratti A, Gellera C, Bosco DA, Bassell GJ, Silani V, Drory VE, Brown RH, Landers JE. Mutations in the profilin 1 gene cause familial amyotrophic lateral sclerosis. Nature 2012;488:499-503.
- Cirulli ET, Lasseigne BN, Petrovski S, Sapp PC, Dion PA, Leblond CS, Couthouis J, Lu YF, Wang Q, Krueger BJ, Ren Z, Keebler J, Han Y, Levy SE, Boone BE, Wimbish JR, Waite LL, Jones AL, Carulli JP, Day-Williams AG, Staropoli JF, Xin WW, Chesi A, Raphael AR, McKenna-Yasek D, Cady J, Vianney de Jong JM, Kenna KP, Smith BN, Topp S, Miller J, Gkazi A, Al-Chalabi A, van den Berg LH, Veldink J, Silani V, Ticozzi N, Shaw CE, Baloh RH, Appel S, Simpson E, Lagier-Tourenne C, Pulst SM, Gibson S, Trojanowski JQ, Elman L, McCluskey L, Grossman M, Shneider NA, Chung WK, Ravits JM, Glass JD, Sims KB, Van Deerlin VM, Maniatis T, Hayes SD, Ordureau A, Swarup S, Landers J, Baas F, Allen AS, Bedlack RS, Harper JW, Gitler AD, Rouleau GA, Brown R, Harms MB, Cooper GM, Harris T, Myers RM, Goldstein DB, FALS Sequencing Consortium. Exome sequencing in amyotrophic lateral sclerosis identifies risk genes and pathways. Science 2015;347:1436-1441.
- Kenna KP, van Doormaal PT, Dekker AM, Ticozzi N, Kenna BJ, Diekstra FP, van Rheenen W, van Eijk KR, Jones AR, Keagle P, Shatunov A, Sproviero W, Smith BN, van Es MA, Topp SD, Kenna A, Miller JW, Fallini C, Tiloca C, McLaughlin RL, Vance C, Troakes C, Colombrita C, Mora G, Calvo A, Verde F, Al-Sarraj S, King A, Calini D, de Belleroche J, Baas F, van der Kooi AJ, de Visser M, Ten Asbroek AL, Sapp PC, McKenna-Yasek D, Polak M, Asress S, Muñoz-Blanco JL, Strom TM, Meitinger T, Morrison KE, Lauria G, Williams KL, Leigh PN, Nicholson GA, Blair IP, Leblond CS, Dion PA, Rouleau GA, Pall H, Shaw PJ, Turner MR, Talbot K, Taroni F, Boylan KB, Van Blitterswijk M, Rademakers R, Esteban-Pérez J, García-Redondo A, Van Damme P, Robberecht W, Chio A, Gellera C, Drepper C, Sendtner M, Ratti A, Glass JD, Mora JS, Basak NA, Hardiman O, Ludolph AC, Andersen PM, Weishaupt JH, Brown RH, Al-Chalabi A, Silani V, Shaw CE, van den Berg LH, Veldink JH, Landers JE, SLAGEN Consortium. NEK1 variants confer susceptibility to amyotrophic lateral sclerosis. Nat Genet 2016;48:1037-1042.
- Chesi A, Staahl BT, Jovičić A, Couthouis J, Fasolino M, Raphael AR, Yamazaki T, Elias L, Polak M, Kelly C, Williams KL, Fifita JA, Maragakis NJ, Nicholson GA, King OD, Reed R, Crabtree GR, Blair IP, Glass JD, Gitler AD. Exome sequencing to identify de novo mutations in sporadic ALS trios. Nat Neurosci 2013;16:851-855.
- Steinberg KM, Yu B, Koboldt DC, Mardis ER, Pamphlett R. Exome sequencing of case-unaffected-parents trios reveals recessive and de novo genetic variants in sporadic ALS. Sci Rep 2015;5:9124.
- Clark JA, Yeaman EJ, Blizzard CA, Chuckowree JA, Dickson TC. A case for microtubule vulnerability in amyotrophic lateral sclerosis: altered dynamics during disease. Front Cell Neurosci 2016;10:204.
- Lim SM, Choi WJ, Oh KW, Xue Y, Choi JY, Kim SH, Nahm M, Kim YE, Lee J, Noh MY, Lee S, Hwang S, Ki CS, Fu XD, Kim SH. Directly converted patient-specific induced neurons mirror the neuropathology of FUS with disrupted nuclear localization in amyotrophic lateral sclerosis. Mol Neurodegener 2016;11:8.
- Heo K, Nahm M, Lee MJ, Kim YE, Ki CS, Kim SH, Lee S. The Rap activator Gef26 regulates synaptic growth and neuronal survival via inhibition of BMP signaling. Mol Brain 2017;10:62.
- Babic M, Russo GJ, Wellington AJ, Sangston RM, Gonzalez M, Zinsmaier KE. Miro's N-terminal GTPase domain is required for transport of mitochondria into axons and dendrites. J Neurosci 2015;35:5754-5771.
- Rana A, Seinen E, Siudeja K, Muntendam R, Srinivasan B, van der Want JJ, Hayflick S, Reijngoud DJ, Kayser O, Sibon OC. Pantethine rescues a Drosophila model for pantothenate kinase-associated neurodegeneration. Proc Natl Acad Sci U S A 2010;107:6988-6993.
- Brooks BR, Miller RG, Swash M, Munsat TL, World Federation of Neurology Research Group on Motor Neuron Diseases. El Escorial revisited: revised criteria for the diagnosis of amyotrophic lateral sclerosis. Amyotroph Lateral Scler Other Motor Neuron Disord 2000;1:293-299.
- Mackenzie IR, Nicholson AM, Sarkar M, Messing J, Purice MD, Pottier C, Annu K, Baker M, Perkerson RB, Kurti A, Matchett BJ, Mittag T, Temirov J, Hsiung GR, Krieger C, Murray ME, Kato M, Fryer JD, Petrucelli L, Zinman L, Weintraub S, Mesulam M, Keith J, Zivkovic SA, Hirsch-Reinshagen V, Roos RP, Züchner S, Graff-Radford NR, Petersen RC, Caselli RJ, Wszolek ZK, Finger E, Lippa C, Lacomis D, Stewart H, Dickson DW, Kim HJ, Rogaeva E, Bigio E, Boylan KB, Taylor JP, Rademakers R. TIA1 mutations in amyotrophic lateral sclerosis and frontotemporal dementia promote phase separation and alter stress granule dynamics. Neuron 2017;95:808-816.e9.
- Molliex A, Temirov J, Lee J, Coughlin M, Kanagaraj AP, Kim HJ, Mittag T, Taylor JP. Phase separation by low complexity domains promotes stress granule assembly and drives pathological fibrillization. Cell 2015;163:123-133.
- Patel A, Lee HO, Jawerth L, Maharana S, Jahnel M, Hein MY, Stoynov S, Mahamid J, Saha S, Franzmann TM, Pozniakovski A, Poser I, Maghelli N, Royer LA, Weigert M, Myers EW, Grill S, Drechsel D, Hyman AA, Alberti S. A liquid-to-solid phase transition of the ALS protein FUS accelerated by disease mutation. Cell 2015;162:1066-1077.
- Webster DR, Borisy GG. Microtubules are acetylated in domains that turn over slowly. J Cell Sci 1989;92:57-65.
- Vadlamudi RK, Barnes CJ, Rayala S, Li F, Balasenthil S, Marcus S, Goodson HV, Sahin AA, Kumar R. p21-activated kinase 1 regulates microtubule dynamics by phosphorylating tubulin cofactor B. Mol Cell Biol 2005;25:3726-3736.
- d'Ydewalle C, Krishnan J, Chiheb DM, Van Damme P, Irobi J, Kozikowski AP, Vanden Berghe P, Timmerman V, Robberecht W, Van Den Bosch L. HDAC6 inhibitors reverse axonal loss in a mouse model of mutant HSPB1-induced Charcot-Marie-Tooth disease. Nat Med 2011;17:968-974.
- Hubbert C, Guardiola A, Shao R, Kawaguchi Y, Ito A, Nixon A, Yoshida M, Wang XF, Yao TP. HDAC6 is a microtubule-associated deacetylase. Nature 2002;417:455-458.
- Roos J, Hummel T, Ng N, Klämbt C, Davis GW. Drosophila Futsch regulates synaptic microtubule organization and is necessary for synaptic growth. Neuron 2000;26:371-382.
- Smiley ST, Reers M, Mottola-Hartshorn C, Lin M, Chen A, Smith TW, Steele GD, Chen LB. Intracellular heterogeneity in mitochondrial membrane potentials revealed by a J-aggregate-forming lipophilic cation JC-1. Proc Natl Acad Sci U S A 1991;88:3671-3675.
- Lindsay J, Esposti MD, Gilmore AP. Bcl-2 proteins and mitochondria--specificity in membrane targeting for death. Biochim Biophys Acta 2011;1813:532-539.
- Smith EF, Shaw PJ, De Vos KJ. The role of mitochondria in amyotrophic lateral sclerosis. Neurosci Lett 2017
- Ferreira A, Cáceres A. The expression of acetylated microtubules during axonal and dendritic growth in cerebellar macroneurons which develop in vitro. Brain Res Dev Brain Res 1989;49:205-213.
- Lim SS, Sammak PJ, Borisy GG. Progressive and spatially differentiated stability of microtubules in developing neuronal cells. J Cell Biol 1989;109:253-263.
- Akella JS, Wloga D, Kim J, Starostina NG, Lyons-Abbott S, Morrissette NS, Dougan ST, Kipreos ET, Gaertig J. MEC-17 is an alpha-tubulin acetyltransferase. Nature 2010;467:218-222.
- Taes I, Timmers M, Hersmus N, Bento-Abreu A, Van Den Bosch L, Van Damme P, Auwerx J, Robberecht W. Hdac6 deletion delays disease progression in the SOD1G93A mouse model of ALS. Hum Mol Genet 2013;22:1783-1790.
- Guo W, Naujock M, Fumagalli L, Vandoorne T, Baatsen P, Boon R, Ordovás L, Patel A, Welters M, Vanwelden T, Geens N, Tricot T, Benoy V, Steyaert J, Lefebvre-Omar C, Boesmans W, Jarpe M, Sterneckert J, Wegner F, Petri S, Bohl D, Vanden Berghe P, Robberecht W, Van Damme P, Verfaillie C, Van Den Bosch L. HDAC6 inhibition reverses axonal transport defects in motor neurons derived from FUS-ALS patients. Nat Commun 2017;8:861.