Articles

Article Tools
 View Full Text View Full Text |
 Abstract Abstract |
 Article as PDF Article as PDF |
 Print this Article Print this Article |
 Pubmed Pubmed |
 PMC PMC |
 PubReader PubReader |
 Export to Citation Export to Citation |
 Email Alerts Email Alerts |
 Open Access Open Access |
Share this article on :



Stats or Metrics
Article
Original Article
Exp Neurobiol 2017; 26(5): 266-277
Published online October 31, 2017
https://doi.org/10.5607/en.2017.26.5.266
© The Korean Society for Brain and Neural Sciences
Beneficial Effects of Silibinin Against Kainic Acidinduced Neurotoxicity in the Hippocampus in vivo
Sehwan Kim1†, Un Ju Jung2†, Yong-Seok Oh3†, Min-Tae Jeon1, Hyung-Jun Kim4, Won-Ho Shin5, Jungwan Hong6* and Sang Ryong Kim1,6*
1School of Life Sciences, BK21 plus KNU Creative BioResearch Group, Kyungpook National University, Daegu 41566, 2Department of Food Science and Nutrition, Pukyong National University, Busan 48513, 3Department of Brain-Cognitive Science, Daegu-Gyeongbuk Institute of Science and Technology, Daegu 42988, 4Department of Neural Development and Disease, Department of Structure & Function of Neural Network, Korea Brain Research Institute, Daegu 41062, 5Predictive Model Research Center, Korea Institute of Toxicology, Daejeon 34114, 6Brain Science and Engineering Institute, Kyungpook National University, Daegu 41944, Korea
Correspondence to: *To whom correspondence should be addressed.
Sang Ryong Kim
TEL: 82-53-950-7362, FAX: 82-53-943-2762, e-mail: srk75@knu.ac.kr
Jungwan Hong
TEL: 82-53-950-7362, FAX: 82-53-943-2762, e-mail: jungwan33@naver.com
†These authors contributed equally to this work.
Silibinin, an active constituent of silymarin extracted from milk thistle, has been previously reported to confer protection to the adult brain against neurodegeneration. However, its effects against epileptic seizures have not been examined yet. In order to investigate the effects of silibinin against epileptic seizures, we used a relevant mouse model in which seizures are manifested as status epilepticus, induced by kainic acid (KA) treatment. Silibinin was injected intraperitoneally, starting 1 day before an intrahippocampal KA injection and continued daily until analysis of each experiment. Our results indicated that silibinin-treatment could reduce seizure susceptibility and frequency of spontaneous recurrent seizures (SRS) induced by KA administration, and attenuate granule cell dispersion (GCD), a morphological alteration characteristic of the dentate gyrus (DG) in temporal lobe epilepsy (TLE). Moreover, its treatment significantly reduced the aberrant levels of apoptotic, autophagic and pro-inflammatory molecules induced by KA administration, resulting in neuroprotection in the hippocampus. Thus, these results suggest that silibinin may be a beneficial natural compound for preventing epileptic events.
Keywords: Silibinin, Epilepsy, Granule cell dispersion, Kainic acid, Neuroprotection
INTRODUCTION
Epilepsy is one of the most common serious neurological disorders characterized by recurrent seizures, and affects around 70 million people worldwide [1]. Temporal lobe epilepsy (TLE) is a type of epilepsy causing focal seizures and its pathophysiological features are hippocampal sclerosis and granule cell dispersion (GCD) [2]. GCD is defined by a scattered distribution of granule cells, extending into the molecular layer of dentate gyrus (DG), and consequent enlargement of the granule cell layer (GCL) in the DG of patients with TLE and animal models of TLE [2,3,4]. Recent studies demonstrated that several molecules involved in the mTORC1 pathway have been implicated in the development of epilepsy [5]. Morphological alteration of GCD caused by modulation of mTORC1 signaling in DG has been also reported by a study [6]. Moreover, a clinical report states that observation of GCD-related cytoarchitectural abnormalities may help in the postsurgical prognosis of TLE patients with DG pathology [7].
Clinical evidence has demonstrated that neuroinflammation is also an important factor in the progression of epilepsy [8]. A recent study showed that activated microglia are observed in the epileptic hippocampus and lead to induction of proinflammatory cytokines such as interleukin-1 beta (IL-1β) and tumor necrosis factor-alpha (TNF-α), causing the neuronal cell death and hyperexcitability involved in epileptogenesis [9]. In addition, it has been reported that hippocampal neuronal cell death, one of the prominent histopathological features in TLE, is usually caused by apoptosis, as well as autophagic stress [10,11]. As mentioned above, autophagy-mediated apoptosis may contribute to the hippocampal neuronal death following seizures, based on apoptotic morphology [12,13].
Current anti-epileptic drugs (AEDs) are effective in managing seizures for only 50~70% of the patients [14] and many AEDs cause some degree of adverse drug reaction, and cause unwanted side effects in some people [15,16,17]. Thus, these reports suggest that natural compounds, which have relatively few side effects compared to synthetic drugs, may be useful as an alternative medicine, and their application, which may have beneficial effects against epileptic seizures, can be used as a novel therapeutic strategy for patients with epilepsy. Recently, it was reported that flavonoids such as naringin, which have anti-oxidative and anti-inflammatory effects against neurodegeneration [18], exhibited neuroprotective effects
MATERIALS AND METHODS
Male C57BL/6 mice (8-week-old, 20~25 g) were obtained from Daehan Biolink (Eumseong, Korea). The mice were housed in a controlled environment and provided with
Injurious seizures were induced by unilateral intrahippocampal KA injection as previously described, with modifications [22]. Briefly, mice were anesthetized using chloral hydrate (360 mg/kg; Sigma, St. Louis, MO, USA), and placed in a stereotaxic frame (David Kopf Instruments, Tujunga, CA, USA). Mice received a unilateral injection of KA [0.2 µg in 2 µl phosphate-buffered saline (PBS); Sigma] into the hippocampus (AP: −2.0 mm; ML: −1.2 mm; DV: -1.5 mm, relative to the bregma; Franklin and Paxinos, 2004) using a 10-µl Hamilton syringe needle (30S) attached to a syringe pump (KD Scientific, NewHope, PA, USA). Mice injected with 2 µl PBS in the same manner were used as controls. After the injection, the needle was left in place for an additional 5 min to limit reflux along the injection track.
Following KA injection, mice were monitored for 6 hr to determine the onset time of seizures. Seizure stages was scored according to a modified scale as previously reported [6]: stage 1, facial movement; stage 2, head nodding and myoclonic twitching; stage 3, forelimb clonus with lordotic posture; stage 4, forelimb clonus with reared posture; and stage 5, tonic-clonic seizure without postural control. In the present study, the mice exhibiting at least stage 3 seizures were classified as positive for seizure onset. To examine the effect of silibinin on chronic seizures, mice were monitored to evaluate the frequency of spontaneous recurrent seizures (SRS) for 108 hr/wk (9 hr/d, 6 d/wk) starting from 3 wk after treatment with KA or silibinin and KA, by video recording as previously described, with some modification [19]. Only the mice exhibiting severe seizures (stages 3–5) were considered for evaluation of SRS. The frequency of SRS during the monitoring period was quantified.
Silibinin [(2R,3R)-3,5,7-trihydroxy-2-[(2R,3R)-3-(4-hydroxy-3-methoxyphenyl)-2-(hydroxymethyl)-2,3-dihydro-1,4-benzodioxin-6-yl]-2,3-dihydrochromen-4-one] is the major active component of silymarin, a standardized extract of milk thistle seeds, which is used as hepatoprotective and anti-cancer agents [20,21]. In order to investigate the effects of silibinin in preventing epileptic seizures, it was freshly prepared by suspension in 0.25% carboxymethylcellulose (Sigma-Aldrich) dissolved in 0.9% saline. Experiments were performed according to the scheme presented in Fig. 1A. For behavioral testing, mice were injected intraperitoneally with silibinin 1 day before KA injection, and then injected with silibinin for a further 2 (latency of seizure) or 35 days (SRS). To determine the effects of silibinin on GCD and neurotoxicity, silibinin treatment was initiated 1 day prior to KA injection, and then continued once daily for 7 days. To examine the expression profiles of protein involved in mTORC1 signaling, apoptosis, autophagy and neuroinflammation, silibinin was injected intraperitoneally daily for 3 days, as with the previous protocols, 1 day prior to KA injection.
Seven days after KA treatment, mice were anesthetized using chloral hydrate (360 mg/kg; Sigma) and perfused transcardially with 4% paraformaldehyde (PFA) in 0.1 M PBS. Brains were immediately removed and placed in the same fixative overnight at 4℃. After post-fixation, the brains ware cryoprotected with 30% sucrose in 0.1 M PBS for 24 hr at 4℃. Serial coronal sections of 30 µm thickness were cut on a freezing microtome (Microm International GmbH, Walldorf, Germany). For Nissl staining, the sections were mounted on gelatin-coated slides and stained with 0.5% cresyl violet (Sigma). The presence of GCD was determined by measuring the average width of the GCL in the mid and medial one-fourth portions of the upper blade of the DG [23]. GCD was quantified as the GCL width on the ipsilateral side and expressed as the percentage of the GCL width relative to the contralateral side.
Cryostat sections were used for immunocytochemistry and immunofluorescence staining using a free-floating procedure, as described previously [19]. Briefly, free-floating sections were washed with 0.1 M PBS and blocked with 0.5% BSA in 0.1 M PBS. Primary antibody incubation was performed two overnight at 4℃. The following primary antibodies were used: anti-neuronal nuclei (NeuN, 1:500, Millipore, Temecula, CA) and anti-ionized calcium-binding adapter molecule 1 (Iba1, 1:2000, Wako Pure Chemical Industries, Japan) and anti-phospho-4E-BP1 (p-4E-BP1, a phosphorylated form of the mTORC1 substrate 4E-BP1; 1:1000; Cell Signaling, Beverly, MA, USA). After repeated washes with PBS, the sections were incubated with biotin-conjugated secondary antibodies, followed by an avidin-biotin complex kit (Vector Laboratories, Burlingame, CA). The signal was detected by incubating the sections in 0.5 mg/ml 3,3′-diaminobenzidine (DAB, Sigma) in 0.1 M PBS containing 0.003% H2O2.
The sections used for immunofluorescence were incubated overnight with one pair of the following antibodies: anti-neuronal nuclei (NeuN, 1:500, Millipore, Temecula, CA), anti-cleaved caspase-3 (c-caspase-3, 1:400, Cell Signaling), anti-cleaved PARP (c-PARP1, 1:400, Cell Signaling), and anti-microtubule-associated protein 1A/1B-light chain 3 (LC3B, 1:200, Cell Signaling). The sections were then incubated with Texas Red- (1:400, Vector Laboratories, Burlingame, CA) and FITC-conjugated IgG (1:200, Jackson ImmunoResearch, West Grove, PA) and then mounted with Vectashield mounting medium (Vector Laboratories, Burlingame, CA). The stained sections were examined under a microscope (Axio Imager; Carl Zeiss, Gottingen, Germany).
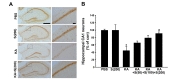
The number of hippocampal CA1 neurons was quantified by staining 30-µm-thick cryosections. Alternate sections were prepared from the coronal brain slice of each animal at 3.3, 3.6, 4.16, and 4.3 mm posterior to the bregma. To maintain consistency across animals, a rectangular box (1×0.05 mm) was centered over the CA1 cell layer beginning 1.5 mm lateral to the midline. Only neurons with visible nuclei were counted by light microscope (Carl Zeiss) at magnification of 400×. The mean number of CA1 neurons in the ipsilateral hippocampus was expressed as a percentage, compared to the contralateral control.
Brain tissues for western blotting were prepared as previously described [19]. DG and CA1 regions of the hippocampus were separated, and each tissue was homogenized, and lysed in buffer containing the protease inhibitor cocktail. Lysates were cleared by centrifugation, protein concentration was determined using a BCA kit (Bio-Rad Laboratories, Hercules, CA, USA). Proteins were separated using gel electrophoresis and transferred onto membranes using an electrophoretic transfer system (Bio-Rad Laboratories). Membranes were incubated overnight at 4℃ with the following antibodies: anti-4E-BP1 (1:1000, Cell Signaling), anti-p-4E-BP1 (1:1000, Cell Signaling), anti-p70 S6 kinase (p70S6K, 1:1000, Cell Signaling), anti-phospho-p70 S6 kinase (p-p70S6K, 1:1000, Cell signaling), anti-β-actin (1:4000, Santa Cruz), anti-LC3B (1:1000, Cell Signaling), anti-c-caspase-3 (1:1000, Cell Signaling), anti-caspase-3 (1:1000, Cell Signaling), anti-tumor necrosis factor alpha (TNF-α, 1:1000, Santa Cruz) and anti-Interleukin 1 beta (IL-1β, 1:1000, Santa Cruz). Membranes were incubated with suitable secondary anti-bodies (Amersham Biosciences, Piscataway, NJ, USA), and blots were developed using enhanced chemiluminescence (ECL) western blotting detection reagents (Amersham Biosciences). For semi-quantitative analyses, band densities were measured using a computerized imaging device and accompanying software (Fuji Film, Tokyo, Japan).
All values are expressed as mean±standard error of the mean (SEM). Multiple comparisons among the groups were performed by one-way analysis of variance (ANOVA) followed by Tukey's
Treatment with KA, an analog of the excitotoxic neurotransmitter glutamate, induces seizures which lead to neuronal death and biochemical changes similar to those observed in human patients with TLE [24]. To evaluate the effects of silibinin administration against KA-induced seizures, mice were injected intraperitoneally with silibinin at various concentrations, starting 1 day before KA injection and continued for 2 days. Silibinin treatment delayed the onset of seizure induced by KA treatment in a dose-dependent manner. Particularly, 200 mg/kg of silibinin significantly delayed seizure onset by 177 min, compared to KA alone (Fig. 1B; *p<0.01
TLE can be characterized by morphological changes associated with broadening of the granule cell layer of the dentate gyrus (DG), termed granule cell dispersion (GCD) [2,3]. It is suggested that restriction of GCD may be useful for preventing and inhibiting epileptic seizures. To investigate whether silibinin attenuates the KA-induced morphological changes of GCD in the DG, brain sections from KA-induced epileptic mice, which are untreated or treated with silibinin, were stained with cresyl violet (Fig. 2A). As we reported previously [6,22], KA treatment induced significant morphological changes such as GCD in the DG compared to PBS-treated controls. However, treatment with various concentrations of silibinin resulted in a dose-dependent reduction of GCD (Fig. 2A and B). Quantitative analysis of GCD showed that KA treatment increased GCL width by 130%, compared to PBS-treated controls (Fig. 2B; *p<0.05
mTOR is a major regulator of cellular changes that mediate epileptogenesis [25], and the hyper-activation of mTOR complex 1 (mTORC1) signaling was observed in dispersed granule cells in a mouse model of TLE and in patients with sclerotic hippocampus [4,26]. In order to investigate whether silibinin administration diminishes KA-induced mTORC1 activation in the DG, we examined the effect of silibinin on phosphorylation of the downstream mTORC1 substrates 4E-BP1 (eukaryotic initiation factor 4E-binding protein 1) and p70S6K1 (p70 ribosomal protein S6 kinase 1). As shown in Fig. 2C, expression of p-4E-BP1 in the DG was increased by KA treatment as compared to PBS-treated controls, and this KA-induced mTORC1 activation was inhibited by silibinin treatment. Immunoblotting results were consistent with this, indicating that silibinin treatment caused a significant reduction of p-4E-BP1 and p-p70S6K levels in the DG which were increased upon KA treatment, compared to KA alone (Fig. 2D and E; #p<0.01 and ##p<0.05
We next investigated the protective effect of silibinin against KA-induced death of hippocampal neurons. Seven days after KA-treatment with daily intraperitoneal injection of various concentrations of silibinin, the surviving neurons in the CA1 region of the hippocampus were detected by NeuN staining (Fig. 3). As shown in Fig. 3A, NeuN staining indicated an apparent neuronal loss in the CA1 region of hippocampus of KA-treated mice, compared to that of PBS-treated mice. However, silibinin treatment prevented the KA-induced neuronal cell loss in the CA1 region of hippocampus. When quantified and expressed as a percentage of the CA1 neurons in the counting area of the ipsilateral hippocampus relative to the contralateral control, the percentage of surviving neurons in the hippocampal CA1 region was significantly lower in the KA-treated mice (45%, *p<0.01
The excitotoxicity mediated by KA could induce neuronal death with apoptotic features in the hippocampus and striatum, resulting from induction of autophagic stress [13,27]. To investigate the protective effects of silibinin against KA-induced apoptosis, we examined the expression of various apoptosis markers, such as cleaved caspase-3 (c-caspase-3) and cleaved PARP-1 (c-PARP-1), in the hippocampus of KA-treated mice at post-lesion 2 days. As shown in Fig. 4A and B, the results of double-immunofluorescence staining demonstrated that numerous c-caspase-3 and c-PARP-1-positive cells were detected in the CA1 of the KA-treated mice, but less c-caspase-3 and c-PARP-1-positive cells were detected in the hippocampus of silibinin-treated mice compared with KA alone. Consistent with the results of double-immunofluorescence staining, western blot analysis revealed that KA treatment induced the significant increases in the levels of c-caspase-3 and c-PARP1 in the hippocampus compared with PBS-treated controls (Fig. 4C and D; *p<0.001 and **p<0.01
Microtubule-associated protein 1A/1B-light chain 3 (LC3) is an important mediator and the most reliable marker of autophagy [13]. LC3-II, required for the formation of autophagosome, is the cleaved and lipidated form of the cytosolic LC3-I [28,29]. To clarify the effects of silibinin against KA-induced autophagic stress, the expression levels of LC3-I and LC3-II were measured by double-immunofluorescence staining (Fig. 4E) and western blot analysis (Fig. 4F and G). Immunofluorescence results showed that KA-treatment could induce an increase in the level of LC3B expression in hippocampal CA1 neurons and its overexpression was inhibited by silibinin treatment (Fig. 4E). In addition, immunoblotting results showed that the levels of LC3-II were significantly increased in mice treated with KA compared to that of PBS-treated controls (Fig. 4F and G; *p<0.001
Recent studies showed that neuroinflammation may play an important role in epilepsy [30]. Moreover, our previous study had reported anti-inflammatory effects of silibinin through inhibition of microglial activation in the animal model of Parkinson's disease [21]. Thus, we further examined whether silibinin contributed to inhibition of microglial activation and reduction of proinflammatory cytokines, such as TNF-α and IL-1β, in the hippocampus of KA-treated mice. Our results showed that KA treatment could induce microglial activation in the hippocampus of mouse brain, and the activation of microglia following KA treatment was inhibited by silibinin treatment (Fig. 5A). Moreover, silibinin treatment significantly inhibited the increase in levels of TNF-α and IL-1β in KA-treated mice, compared to KA alone (Fig. 5B and C; #p<0.001 and ##p<0.05
Epilepsy is a common neurological disorder characterized by epileptic seizures [31]. Over the last two decades, medications available for epilepsy have improved substantially. However, treatments for epilepsy are based on drugs that only alleviate the symptoms, despite efforts to identify more efficient therapeutic agents for epilepsy [32]. To overcome these limitations, recent studies have focused on the neuroprotective actions of flavonoids against injury induced by neurotoxins within the brain, including an ability to suppress neuroinflammation and the potential to promote memory [33]. Recently, silibinin, a flavonoid derived from milk thistle seeds, has been reported to inhibit amyloid β peptide aggregation and acetylcholinesterase activity by reducing oxidative stress and inflammation in animal models of Alzheimer's disease (AD) [34,35]. We have also previously revealed anti-inflammatory and neuroprotective effects of silibinin as a consequence of the inhibition of iNOS and proinflammatory cytokines, such as TNF-α and IL-1β, in a rat model of Parkinson's disease [21]. Taken together, these results suggest beneficial effects of silibinin in various brain diseases, including epilepsy. In the present study, our results showed that silibinin treatment delayed the onset of KA-induced seizures. Furthermore, silibinin significantly reduced occurrence of SRS in KA-treated mice (Fig. 1). These results indicate that silibinin may have therapeutic value as an antiepileptic drug.
Since GCD is observed in patients with epilepsy, studies in animal models suggest that GCD may be a consequence of enhanced proliferation of granule cell precursors triggered by seizures [36]. Pathophysiological aspects of epilepsy such as GCD were influenced by abnormal hyperactivation of mTORC1 in the hippocampus of animal models of epilepsy [4,26]. As described above, silibinin has been shown to have potential antiepileptic properties against KA-induced seizures. However, it was unclear how silibinin could affect pathophysiological changes induced by KA, such as GCD and neuronal cell death that lead to consequent development of epileptic seizures [37,38]. In this study, our results showed that silibinin administration dramatically inhibited KA-induced GCD and mTORC1 activation. This was confirmed through immunoblotting to assess levels of the phosphorylated forms of downstream mTORC1 substrates 4E-BP1 and p70S6K (Fig. 2).
Neuroprotective effects of silibinin have been recently reported in animal models of Alzheimer's disease, in which systemic administration of silibinin increased the concentration of BDNF by reducing the Aβ level [34]. In addition, we demonstrated neuroprotective effect of silibinin in the substantia nigra, which was associated with the inhibition of inflammatory cytokine production in animal models of Parkinson's disease [21]. In this study, we demonstrated that treatment with silibinin protected the hippocampal CA1 neurons from KA-induced neuronal cell death (Fig. 3). Also, silibinin is beneficial for treating epilepsy through its ability to inhibit GCD
The current studies depend on the fact that TLE characterized by excitotoxic cell death is commonly modeled in rodents by KA treatment [39]. KA-induced excitotoxicity increased the expression of proteins mediated by apoptosis or autophagy [13]. Also, several recent studies have reported that autophagic stress induces neuronal death in animal models of neurodegenerative disease [13,40,41]. These evidences suggest that the control of KA-induced apoptosis and autophagy may be important to protect the hippocampal neurons. In the present study, we found that KA treatment elicited an increase of caspase-3 activation and PARP-1 cleavage in the hippocampus CA1 neurons. However, silibinin treatment showed dramatic inhibition in both caspase-3 activation and PARP1 cleavage induced by KA treatment. In addition, recent reports have shown that hyperactivated mTORC1 accelerates apoptosis in AD patients [42,43]. Several studies have also revealed that inhibition of mTORC1 by rapamycin prevents glutaminolysis and oxidative stress-induced apoptosis in cancer cells [44,45]. Taken together, our results suggest that the anti-apoptotic effects of silibinin could be associated with a suppression of mTORC1 signaling. Similar to the results in apoptosis, LC3B expression and LC3-II levels, as a marker of autophagy, significantly increased in the hippocampus after KA treatment and significant inhibition of LC3B and LC3-II were observed after silibinin administration (Fig. 4). Collectively, these results suggest that the anti-apoptotic and antiautophagic properties of silibinin may contribute to the neuroprotective effects of silibinin observed in a KA-induced excitotoxicity animal model.
Inflammation is recognized as a feature of acute and chronic neurological disorders. However, the function of inflammation in the pathogenesis of epilepsy and seizure-induced brain damage has been appreciated only recently [46]. The effect of silibinin on neuroinflammation in KA-induced excitotoxicity animal model is not well-understood. Thus, we examined the change in levels of inflammatory cytokines caused by silibinin treatment in hippocampus of KA-treated mice. In the present study, our results indicated that treatment with silibinin prevented microglial activation induced by KA treatment, resulting in the significant inhibition of expressions of TNF-α and IL-1β in the KA-treated hippocampus (Fig. 5). These findings suggest that silibinin affords strong protection against KA-induced excitotoxic cell death via prevention of activated microglia-derived neuroinflammation in the KA-treated hippocampus.
In conclusion, we have demonstrated the beneficial effects of silibinin against KA-induced excitotoxicity in the animal model of epilepsy. Our results demonstrate that treatment with silibinin could significantly decrease seizure susceptibility in KA-treated mice. The anti-epileptic effects of silibinin are mediated by the inhibition of GCD

{kind=link}

{kind=link}
{kind=link}

{kind=link}

{kind=link}
- Gourie-Devi M, Gururaj G, Satishchandra P, Subbakrishna DK. Prevalence of neurological disorders in Bangalore, India: a community-based study with a comparison between urban and rural areas. Neuroepidemiology 2004;23:261-268.

- Houser CR. Granule cell dispersion in the dentate gyrus of humans with temporal lobe epilepsy. Brain Res 1990;535:195-204.
- Marucci G, Rubboli G, Giulioni M. Role of dentate gyrus alterations in mesial temporal sclerosis. Clin Neuropathol 2010;29:32-35.
- Shima A, Nitta N, Suzuki F, Laharie AM, Nozaki K, Depaulis A. Activation of mTOR signaling pathway is secondary to neuronal excitability in a mouse model of mesio-temporal lobe epilepsy. Eur J Neurosci 2015;41:976-988.
- Ostendorf AP, Wong M. mTOR inhibition in epilepsy: rationale and clinical perspectives. CNS Drugs 2015;29:91-99.
- Park J, Jeong KH, Shin WH, Bae YS, Jung UJ, Kim SR. Naringenin ameliorates kainic acid-induced morphological alterations in the dentate gyrus in a mouse model of temporal lobe epilepsy. Neuroreport 2016;27:1182-1189.
- Na M, Liu Y, Shi C, Gao W, Ge H, Wang Y, Wang H, Long Y, Shen H, Shi C, Lin Z. Prognostic value of CA4/DG volumetry with 3T magnetic resonance imaging on postoperative outcome of epilepsy patients with dentate gyrus pathology. Epilepsy Res 2014;108:1315-1325.
- Vezzani A, French J, Bartfai T, Baram TZ. The role of inflammation in epilepsy. Nat Rev Neurol 2011;7:31-40.
- Dey A, Kang X, Qiu J, Du Y, Jiang J. Anti-inflammatory small molecules to treat seizures and epilepsy: from bench to bedside. Trends Pharmacol Sci 2016;37:463-484.
- Rocha LL, Lopez-Meraz ML, Niquet J, Wasterlain CG. Do single seizures cause neuronal death in the human hippocampus?. Epilepsy Curr 2007;7:77-81.
- Nixon RA, Yang DS. Autophagy and neuronal cell death in neurological disorders. Cold Spring Harb Perspect Biol 2012;4:a008839.
- Meller R, Schindler CK, Chu XP, Xiong ZG, Cameron JA, Simon RP, Henshall DC. Seizure-like activity leads to the release of BAD from 14-3-3 protein and cell death in hippocampal neurons in vitro. Cell Death Differ 2003;10:539-547.
- Wang Y, Han R, Liang ZQ, Wu JC, Zhang XD, Gu ZL, Qin ZH. An autophagic mechanism is involved in apoptotic death of rat striatal neurons induced by the non-N-methyl-D-aspartate receptor agonist kainic acid. Autophagy 2008;4:214-226.
- Khodayar MJ, Salehi S, Rezaei M, Siahpoosh A, Khazaei A, Houshmand G. Evaluation of the effect of naringenin on pentylenetetrazole and maximal electroshock-induced convulsions in mice. Jundishapur J Nat Pharm Prod 2017;12:e31384.
- Błaszczyk B, Szpringer M, Czuczwar SJ, Lasoń W. Single centre 20 year survey of antiepileptic drug-induced hypersensitivity reactions. Pharmacol Rep 2013;65:399-409.
- Then SM, Rani ZZ, Raymond AA, Ratnaningrum S, Jamal R. Frequency of the HLA-B*1502 allele contributing to carbamazepine-induced hypersensitivity reactions in a cohort of Malaysian epilepsy patients. Asian Pac J Allergy Immunol 2011;29:290-293.
- Pirmohamed M, Friedmann PS, Molokhia M, Loke YK, Smith C, Phillips E, La Grenade L, Carleton B, Papaluca-Amati M, Demoly P, Shear NH. Phenotype standardization for immune-mediated drug-induced skin injury. Clin Pharmacol Ther 2011;89:896-901.
- Gopinath K, Sudhandiran G. Naringin modulates oxidative stress and inflammation in 3-nitropropionic acidinduced neurodegeneration through the activation of nuclear factor-erythroid 2-related factor-2 signalling pathway. Neuroscience 2012;227:134-143.
- Jeong KH, Jung UJ, Kim SR. Naringin attenuates autophagic stress and neuroinflammation in kainic acid-treated hippocampus in vivo. Evid Based Complement Alternat Med 2015;2015:354326.
- Jangra A, Kasbe P, Pandey SN, Dwivedi S, Gurjar SS, Kwatra M, Mishra M, Venu AK, Sulakhiya K, Gogoi R, Sarma N, Bezbaruah BK, Lahkar M. Hesperidin and silibinin ameliorate aluminum-induced neurotoxicity: modulation of antioxidants and inflammatory cytokines level in mice hippocampus. Biol Trace Elem Res 2015;168:462-471.
- Jung UJ, Jeon MT, Choi MS, Kim SR. Silibinin attenuates MPP+-induced neurotoxicity in the substantia nigra in vivo. J Med Food 2014;17:599-605.
- Jang H, Jeong KH, Kim SR. Naringin attenuates granule cell dispersion in the dentate gyrus in a mouse model of temporal lobe epilepsy. Epilepsy Res 2016;123:6-10.
- Suzuki F, Junier MP, Guilhem D, Sørensen JC, Onteniente B. Morphogenetic effect of kainate on adult hippocampal neurons associated with a prolonged expression of brain-derived neurotrophic factor. Neuroscience 1995;64:665-674.
- Baran H, Hörtnagl H, Hornykiewicz O. Kainic acid-induced seizures: potentiation by α-methyl-p-tyrosine. Brain Res 1989;495:253-260.
- Meng XF, Yu JT, Song JH, Chi S, Tan L. Role of the mTOR signaling pathway in epilepsy. J Neurol Sci 2013;332:4-15.
- Sha LZ, Xing XL, Zhang D, Yao Y, Dou WC, Jin LR, Wu LW, Xu Q. Mapping the spatio-temporal pattern of the mammalian target of rapamycin (mTOR) activation in temporal lobe epilepsy. PLoS One 2012;7:e39152.
- Weiss S, Cataltepe O, Cole AJ. Anatomical studies of DNA fragmentation in rat brain after systemic kainate administration. Neuroscience 1996;74:541-551.
- Tanida I, Ueno T, Kominami E. LC3 conjugation system in mammalian autophagy. Int J Biochem Cell Biol 2004;36:2503-2518.
- Kabeya Y, Mizushima N, Ueno T, Yamamoto A, Kirisako T, Noda T, Kominami E, Ohsumi Y, Yoshimori T. LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J 2000;19:5720-5728.
- Vezzani A, Moneta D, Richichi C, Aliprandi M, Burrows SJ, Ravizza T, Perego C, De Simoni MG. Functional role of inflammatory cytokines and antiinflammatory molecules in seizures and epileptogenesis. Epilepsia 2002;43:30-35.
- Chang BS, Lowenstein DH. Epilepsy. N Engl J Med 2003;349:1257-1266.
- Grosso C, Valentão P, Ferreres F, Andrade PB. The use of flavonoids in central nervous system disorders. Curr Med Chem 2013;20:4694-4719.
- Spencer JP. Flavonoids and brain health: multiple effects underpinned by common mechanisms. Genes Nutr 2009;4:243-250.
- Duan S, Guan X, Lin R, Liu X, Yan Y, Lin R, Zhang T, Chen X, Huang J, Sun X, Li Q, Fang S, Xu J, Yao Z, Gu H. Silibinin inhibits acetylcholinesterase activity and amyloid β peptide aggregation: a dual-target drug for the treatment of Alzheimer's disease. Neurobiol Aging 2015;36:1792-1807.
- Lu P, Mamiya T, Lu LL, Mouri A, Zou L, Nagai T, Hiramatsu M, Ikejima T, Nabeshima T. Silibinin prevents amyloid beta peptide-induced memory impairment and oxidative stress in mice. Br J Pharmacol 2009;157:1270-1277.
- Thom M, Martinian L, Williams G, Stoeber K, Sisodiya SM. Cell proliferation and granule cell dispersion in human hippocampal sclerosis. J Neuropathol Exp Neurol 2005;64:194-201.
- Wang Q, Yu S, Simonyi A, Sun GY, Sun AY. Kainic acid-mediated excitotoxicity as a model for neurodegeneration. Mol Neurobiol 2005;31:3-16.
- Jin Y, Lim CM, Kim SW, Park JY, Seo JS, Han PL, Yoon SH, Lee JK. Fluoxetine attenuates kainic acid-induced neuronal cell death in the mouse hippocampus. Brain Res 2009;1281:108-116.
- Zhu ZQ, Armstrong DL, Hamilton WJ, Grossman RG. Disproportionate loss of CA4 parvalbumin-immunoreactive interneurons in patients with Ammon’s horn sclerosis. J Neuropathol Exp Neurol 1997;56:988-998.
- Lum JJ, Bauer DE, Kong M, Harris MH, Li C, Lindsten T, Thompson CB. Growth factor regulation of autophagy and cell survival in the absence of apoptosis. Cell 2005;120:237-248.
- Boya P, González-Polo RA, Casares N, Perfettini JL, Dessen P, Larochette N, Métivier D, Meley D, Souquere S, Yoshimori T, Pierron G, Codogno P, Kroemer G. Inhibition of macroautophagy triggers apoptosis. Mol Cell Biol 2005;25:1025-1040.
- Lee KH, Lee SJ, Lee HJ, Choi GE, Jung YH, Kim DI, Gabr AA, Ryu JM, Han HJ. Amyloid β1-42 (Aβ1-42) induces the CDK2-mediated phosphorylation of Tau through the activation of the mTORC1 signaling pathway while promoting neuronal cell death. Front Mol Neurosci 2017;10:229.
- Son JH, Shim JH, Kim KH, Ha JY, Han JY. Neuronal autophagy and neurodegenerative diseases. Exp Mol Med 2012;44:89-98.
- Villar VH, Nguyen TL, Delcroix V, Terés S, Bouchecareilh M, Salin B, Bodineau C, Vacher P, Priault M, Soubeyran P, Durán RV. mTORC1 inhibition in cancer cells protects from glutaminolysis-mediated apoptosis during nutrient limitation. Nat Commun 2017;8:14124.
- Thedieck K, Holzwarth B, Prentzell MT, Boehlke C, Kläsener K, Ruf S, Sonntag AG, Maerz L, Grellscheid SN, Kremmer E, Nitschke R, Kuehn EW, Jonker JW, Groen AK, Reth M, Hall MN, Baumeister R. Inhibition of mTORC1 by astrin and stress granules prevents apoptosis in cancer cells. Cell 2013;154:859-874.
- Golechha M, Chaudhry U, Bhatia J, Saluja D, Arya DS. Naringin protects against kainic acid-induced status epilepticus in rats: evidence for an antioxidant, anti-inflammatory and neuroprotective intervention. Biol Pharm Bull 2011;34:360-365.