Articles

Article Tools
 View Full Text View Full Text |
 Abstract Abstract |
 Article as PDF Article as PDF |
 Print this Article Print this Article |
 Pubmed Pubmed |
 PMC PMC |
 PubReader PubReader |
 Export to Citation Export to Citation |
 Email Alerts Email Alerts |
 Open Access Open Access |
Share this article on :



Stats or Metrics
Article
Original Article
Exp Neurobiol 2018; 27(3): 226-237
Published online June 30, 2018
https://doi.org/10.5607/en.2018.27.3.226
© The Korean Society for Brain and Neural Sciences
Morin Prevents Granule Cell Dispersion and Neurotoxicity via Suppression of mTORC1 in a Kainic Acid-induced Seizure Model
Ji Min Lee1†, Jungwan Hong2†, Gyeong Joon Moon1†, Un Ju Jung3, So-Yoon Won4 and Sang Ryong Kim1,2*
1School of Life Sciences, BK21 plus KNU Creative BioResearch Group, Kyungpook National University, Daegu 41566, 2Brain Science and Engineering Institute, Kyungpook National University, Daegu 41944, 3Department of Food Science and Nutrition, Pukyong National University, Busan 48513, 4Department of Biochemistry and Medical Research Center, Chungbuk National University, Cheongju 28644, Korea
Correspondence to: *To whom correspondence should be addressed.
TEL: 82-53-950-7362, FAX: 82-53-943-2762
e-mail: srk75@knu.ac.kr
†These authors contributed equally.
Abstract
An abnormal reorganization of the dentate gyrus and neurotoxic events are important phenotypes in the hippocampus of patients with temporal lobe epilepsy (TLE). The effects of morin, a bioflavonoid constituent of many herbs and fruits, on epileptic seizures have not yet been elucidated, though its beneficial effects, such as its anti-inflammatory and neuroprotective properties, are well-described in various neurodegenerative diseases. In the present study, we investigated whether treatment with morin hydrate (MH) can reduce the susceptibility to seizures, granule cell dispersion (GCD), mammalian target of rapamycin complex 1 (mTORC1) activity, and the increases in the levels of apoptotic molecules and inflammatory cytokines in the kainic acid (KA)-induced seizure mouse model. Our results showed that oral administration of MH could reduce susceptibility to seizures and lead to the inhibition of GCD and mTORC1 activity in the KA-treated hippocampus. Moreover, treatment with MH significantly reduced the increased levels of apoptotic signaling molecules and pro-inflammatory mediators in the KA-treated hippocampus compared with control mice, suggesting a neuroprotective role. Therefore, these results suggest that morin has a therapeutic potential against epilepsy through its abilities to inhibit GCD and neurotoxic events in the
Graphical Abstract
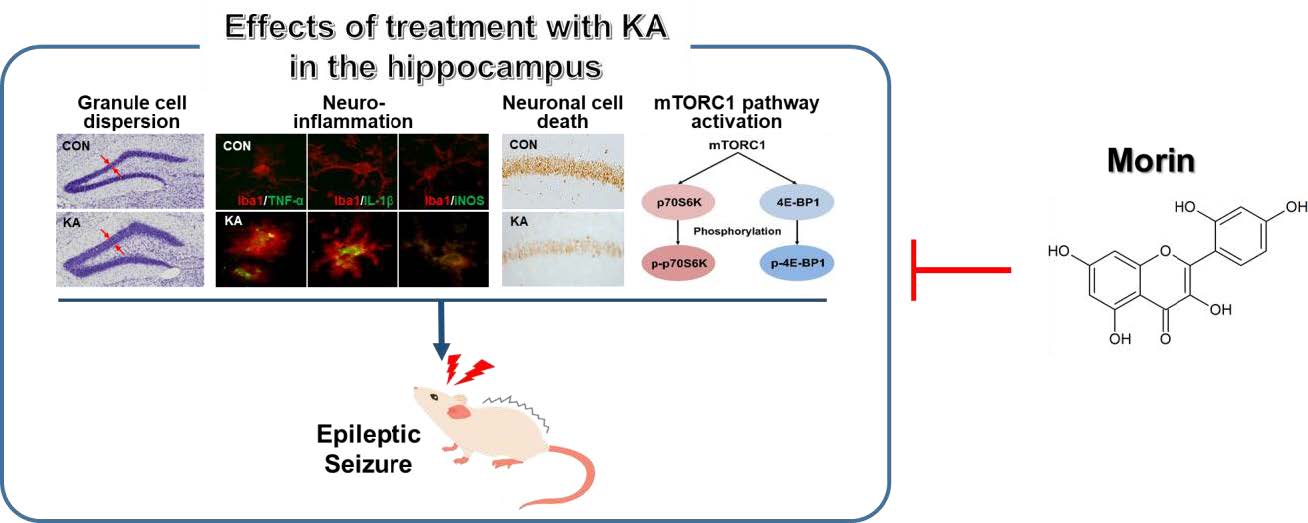
Keywords: Morin, Seizure, Granule cell dispersion, Kainic acid, Neuroprotection
INTRODUCTION
Epilepsy, a serious neurological disease, causes unprovoked, recurrent seizures [1]. Temporal lobe epilepsy (TLE) is the most common form of epilepsy and is characterized by neuronal cell loss and granule cell dispersion (GCD), a structural abnormality of the dentate gyrus (DG) present in approximately 50% of patients with TLE [2]. GCD involves the enlargement of the granule cell layer (GCL) and loss of a clear outer boundary between the molecular and granular layers in the DG [3,4,5]. Recent studies have also shown morphological changes in granule cells and mossy fiber sprouting in the DG. Additionally, GCD-associated activation of mTORC1 was observed in the hippocampus of TLE patients and animal models [6,7,8,9].
Neuronal death and injury are observed in various models of epilepsy and are considered a prerequisite to seizure-induced epilepsy [10]. Recently, many studies demonstrated that neurotoxic inflammation in the brain could increase the predisposition to seizures and be associated with alterations in seizure-induced neuropathology and neuronal excitability [11,12]. In addition, apoptosis may be associated with seizures, resulting in the loss of hippocampal neurons [13,14]. The administration of kainic acid (KA) is widely used as a model of TLE and induces behavioral symptoms and pathological changes similar to human TLE [7,15,16,17].
There has been a growing interest in exploiting natural, pure compounds due to their affordability, minimal side effects and beneficial effects as alternatives to traditional medicine. Their neuroprotective effects against excitotoxicity may be of particular therapeutic value for brain diseases [18,19]. Morin is a bioflavonoid isolated as a yellowish pigment from
MATERIALS AND METHODS
Male C57BL/6 mice (age: 8 weeks, body weight: 22~23 g) were purchased from Daehan Biolink (Eumseong, Korea) and maintained for 1 week before experiments. The mice were housed in a controlled environment and provided with food and water
Seizures were induced by a unilateral injection of KA in the hippocampus of mice brain as previously described with minor modifications [7]. Briefly, mice were positioned on a stereotaxic frame (David Kopf Instruments, Tujunga, CA, USA) after induction of anesthesia. Each mouse received an unilateral injection of KA [0.2 µg in 2 µl phosphate-buffered saline (PBS); Sigma] using a 10 µl Hamilton syringe (30-S needle) attached to a syringe pump (KD Scientific, New Hope, PA, USA) into the hippocampus (AP: −2.0 mm; ML: −1.2 mm; DV: −1.5 mm, relative to the bregma). Mice injected with 2 µl of PBS alone were used as controls. After the injection, the needle was left in place for an additional 5 minutes to limit reflux along the injection track.
To investigate the effects of MH [2′,3,4′,5,7-pentahydroxyflavone] (Fig. 1A) in preventing epileptic seizures, the drug was freshly prepared in suspension of 0.01 M PBS. Experiments were performed according to the scheme presented in Fig. 1B. Mice were administered orally with MH in various doses (20, 40, or 80 mg/10 ml/kg per day; Sigma) 1 hour and 1 day before the KA injection to measure seizure onset. To analysis the occurrence of spontaneous recurrent seizures (SRS) and the population of mossy fiber sprouting, MH was orally administered to the mice, one times daily from 2 days before the KA injection to 35 days. To study the effects of MH on properties such as the inhibition of GCD and neurotoxicity, the drug was initiated 1 day before the KA injection, then continued for 7 days. For experiments assessing the protein levels of mammalian target of rapamycin complex 1 (mTORC1), neuroinflammation and apoptosis, mice were treated with MH for 3 days and sacrificed 2 days after KA treatment.
After KA treatment, mice were monitored for up to 3 hours in order to evaluate the time of seizure onset. The severity of seizures was classified into five stages according to a modified scale as previously described [8,27,28]. The stages were: stage 1, characterized by facial movements; stage 2, characterized by head nodding and myoclonic twitching; stage 3, characterized by forelimb clonus with lordotic posture; stage 4, characterized by forelimb clonus with reared posture; and stage 5, characterized by tonic-clonic seizures without postural control. In the present study, mice exhibiting seizures with at least stage 3 severity were considered positive for seizure onset. In addition, 3 weeks after KA treatment, the frequency of SRS was measured over a period of two weeks [54 h/week (9 h/day, 6 days/week)] using video recording, as previously described, with some modification [29].
Seven days after KA treatment, mice were sacrificed and subjected to transcardial perfusion with 4% paraformaldehyde in 0.1 M PBS. The brains were obtained and post-fixed overnight at 4℃ and then cut in 30-µm-thick coronal sections on a HM525 NX cryostat (Thermo Scientific, Germany). For Nissl staining, the sections were mounted on gelatin-coated slides and stained with 0.5% cresyl violet (Sigma). The presence of GCD was determined by measuring the average width of the GCL in the mid and medial quarter portions of the upper blade of the DG [29]. GCD was expressed as a percentage of the GCL width relative to the contralateral side.
Brain sections were mounted on gelatin-coated slides and subsequently treated as previously described [30,31,32], using Timm's staining to reveal mossy fibers. Briefly, the slides were placed in the staining solution containing a 6:1:3 mixture of gum arabic (50%, w/v), 2 M citric acid-sodium citrate buffer, 0.5 M hydroquinone, and with 0.25 ml of a silver nitrate solution (17%, w/v). The developing process was monitored periodically over a period of 3 hours until the desired level of staining was attained. The sections were rinsed in water for 15 min, dehydrated with ethanol, and cleared in xylene before being cover slipped. To semi-qualitatively assess mossy fiber sprouting, sections at equivalent positions relative to bregma ipsilateral and contralateral to injury were examined and assigned Timm scores ranging from 0~3, with a score of 0 corresponding to little to no granular staining, 1 indicating moderate Timm staining through the granule cell layer, but not into the inner molecular layer, 2 indicating continuous staining through the granule cell layer with discontinuous puncta in the inner molecular layer, and 3 indicting a continuous band of staining in the inner molecular layer. The scorer was blinded to treatment [30,31].
Brain sections were prepared for immunohistochemistry and immunofluorescence staining as previously described [7]. Briefly, free-floating sections were washed with 0.1 M PBS and blocked with 0.5% bovine serum albumin in 0.1 M PBS. Primary antibody incubation was performed two overnight at 4℃. The following primary antibodies were used: anti-neuronal nuclei (NeuN; 1:500; Millipore, Temecula, CA, USA) and anti-phospho-4E-BP1 [p-4E-BP1, a phosphorylated form of eukaryotic initiation factor 4E-binding protein 1 (4E-BP1); 1:1000; Cell Signaling, Beverly, MA, USA]. The sections were subsequently washed and incubated with biotin-conjugated secondary antibodies, followed by an avidinbiotin complex kit (Vector Laboratories, Burlingame, CA, USA). The signal was visualized using 3,3′-diaminobenzidine (0.5 mg/ml; Sigma) with hydrogen peroxide (0.003%; Sigma).
The sections used for immunofluorescence were incubated two overnight with one pair of the following antibodies: anti-NeuN (1:500; Millipore), anti-cleaved caspase-3 (c-caspase-3; 1:400; Cell Signaling), anti-cleaved Poly [ADP-ribose] polymeras-1 (PARP-1) (c-PARP-1; 1:400; Cell Signaling), anti-ionized calcium-binding adapter molecule 1 (Iba1; 1:500; Wako Pure Chemical Industries, Japan), anti-Interleukin 1 beta (IL-1β; 1:500; Santa Cruz), anti-tumor necrosis factor alpha (TNF-α; 1:1000; Santa Cruz) and anti-inducible nitric oxide synthase (iNOS; 1:200; Abcam, Cambridge, MN, USA). The sections were then incubated with Texas Red-conjugated IgG (1:400; Vector Laboratories) and fluorescein isothiocyanate-conjugated IgG (1:200; Jackson ImmunoResearch, West Grove, PA, USA), and mounted with Vectashield mounting medium (Vector Laboratories). The stained sections were examined under a microscope (Axio Imager; Carl Zeiss, Gottingen, Germany).
The number of hippocampal CA1 neurons was quantified as previously described with some modifications [33]. All quantitative analyses were performed in a blind manner. Alternate sections were prepared from the coronal brain slice of each animal at 1.7, 1.8, 2.0, and 2.1 mm posterior to bregma. To ensure consistency in tissue sampling, a rectangular box (1×0.05 mm) was centered over the CA1 cell layer beginning 1.0 mm lateral to the midline. Only neurons with visible nuclei were counted using a light microscope (Carl Zeiss) at a magnification of 200×. The mean number of CA1 neurons in the ipsilateral hippocampus was expressed as a percentage, compared with the contralateral control.
Hippocampus were dissected from brain tissue and used for western blotting, as described previously [7,34]. Briefly, each tissue was homogenized in lysis buffer containing a protease inhibitor cocktail (Sigma) and cleared by centrifugation. The protein concentration was determined using a BCA assay kit (Bio-Rad Laboratories, Hercules, CA, USA). Proteins were separated by gel electrophoresis and transferred to polyvinylidene difluoride (PVDF) membranes (Bio-Rad Laboratories). The PVDF membranes were incubated overnight at 4℃ with the following antibodies: anti-4E-BP1 (1:1000, Cell signaling), anti-p-4E-BP1 (1:1000, Cell signaling), anti-p70S6 kinase (p70S6K, 1:1000, Cell Signaling), anti-phospho-p70S6K (p-p70S6K, 1:1000, Cell signaling), anti-β-actin (1:4000, Santa Cruz), anti-caspase-3 (1:1000, Cell Signaling), anti-c-caspase-3 (1:1000, Cell Signaling), anti-PARP-1 (1:1000, Cell signaling), anti-c-PARP-1 (1:1000, Cell signaling), anti-TNF-α (1:1000, Santa Cruz), anti-IL-1β (1:1000, Santa Cruz), and anti-iNOS (1:500, Abcam). The membranes were subsequently incubated with secondary antibodies (GE Healthcare, Piscataway, NJ, USA), and blots were developed using enhanced chemiluminescence western blotting detection reagents (GE Healthcare). For semi-quantitative analyses, band densities were measured using a computer imaging device and accompanying software (Fuji Film, Tokyo, Japan).
All values are expressed as mean±standard error of the mean (SEM). The Shapiro-Wilk test was used to confirm normal distribution prior to statistical analyses. Differences between the two groups were analyzed with either the Student's t-test or Mann-Whitney U test. Multiple comparisons among groups were performed using either the one-way analysis of variance (ANOVA) or Kruskal-Wallis test, followed by Tukey's
RESULTS
KA-induced excitotoxicity is established to induce seizures; behavioral changes; and neuro-pathophysiological features, such as GCD, similar to those found in patients with TLE [2,7]. The hyperactivation of mTORC1 was also observed in dispersed granule cells in a mouse model of TLE and in patients with a sclerotic hippocampus [6]. To investigate the effects of MH on KA-induced seizures, various doses of MH (20, 40, or 80 mg/10 ml/kg per day) were orally administrated 1 hour and 1 day before the KA injection. The effect of MH in delaying seizures was dose-dependent. 80 mg/kg of MH significantly delayed the onset time of seizures compared to KA alone within 3 hours after KA treatment (Fig. 1C; **p<0.01 vs. KA-treated mice). Furthermore, we investigated whether oral administration of MH could repress the occurrence of SRS after KA treatment. To measure SRS, mice received 80 mg/kg of MH 1 hour and 1 day before KA injection. This continued for 5 weeks. The results are shown in Fig. 1C. The frequency of SRS tended to decrease in the MH-treated mice compared to mice which received KA alone (Fig. 1D, p=0.163 vs. KA-treated mice).
To evaluate whether the administration of MH could diminish KA-induced morphological changes in GCD and mossy fiber sprouting in the DG, we further measured GCD and mossy fiber sprouting using Nissl staining and Timm's staining, respectively, after a latency period of 7 days. The results of Nissl staining revealed that a unilateral injection of KA into the hippocampus induced GCD in the DG, but not in the DG of single MH-treated mice (Fig. 2A). However, treatment with MH resulted in a dose-dependent reduction of GCD in KA-treated hippocampus (Fig. 2A and B). The alterations in GCD were quantified by comparing the experimental sides with the contralateral sides. KA treatment led to a significant increase in GCD by 143%, compared to controls (Fig. 2B; **p<0.01). Notably, administration of 40 and 80 mg/kg of MH significantly inhibited the formation of GCD caused by KA treatment, compared with KA alone (Fig. 2B; $p<0.05 and #p<0.05, respectively, vs. KA alone). Furthermore, Timm-positive staining showed that the aberrant mossy fiber sprouting was significantly higher in the GCL and inner molecular layer, where mossy fibers project in the KA-treated mice than in controls (Fig. 2C and D; p<0.001). In contrast, MH-treated mice for 5 weeks showed a decreasing tendency of mossy fiber sprouting in the DG of KA-treated hippocampus (Fig. 2C and D; p=0.088).
Several previous reports indicated that KA-induced status epilepticus results in increases in mTOR activity in adult animals [35,36,37,38,39]. The use of mTOR inhibitors, such as rapamycin, were also suggested as a rational and efficacious strategy for preventing epilepsy [9]. To examine whether MH treatment can prevent KA-induced activation of mTORC1 in the DG, we measured the levels of p-4E-BP1, an mTORC1 substrate, in the DG of mice brains at early time points. Mice were pretreated with 80 mg/kg of MH 1 hour and 1 day prior to KA treatment. Subsequently, one group of mice was sacrificed 30 min and 90 min after KA injection. The activation of mTORC1 was assessed by measuring phosphorylation of p70S6K in the DG using western blot analysis (Fig. 3A). Our results showed that phosphorylation of p70S6K significantly increased in the DG at early time points (both 30 min and 90 min) after KA injection compared to controls (Fig. 3B; **p<0.01, ***p<0.001 vs. CON, respectively), and MH-treated mice showed a decreasing trend in the levels of p-p70S6K at 30 min after KA-treated hippocampus (Fig. 3B; p=0.144). Moreover, after 90 min KA-treatment, we found a significant reduction of p-p70S6K in MH-treated mice compared to KA alone-treated mice (Fig. 3B; #p<0.05 vs. KA alone). In addition, to confirm whether continuous administration of MH can inhibit the activation of mTORC1 following KA-treatment, mice received MH for 2 more days after KA-treatment. Our immunohistochemical results showed that KA induced phosphorylation of 4E-BP1 in the DG, and MH-treatment effectively inhibited the p-4E-BP1 in the KA-treated hippocampus (Fig. 3C). Similar to these results, KA treatment induced a significant increase in the levels of p-4E-BP1 and p-p70S6K 2 days after seizure induction (Fig. 3D, E; ***p<0.001 vs. CON and 80 mg/kg MH-treated mice, respectively). However, treatment with MH following KA administration led to a significant reduction in the levels of p-4E-BP1 and p-p70S6K compared to KA alone (Fig. 3D, E; #p<0.05).
To investigate the protective effect of morin against KA-induced neurotoxicity in the hippocampus, 80 mg/kg of MH was orally administered 1 day before the KA injection, then continued for 7 days. As shown in Fig. 4A, an intra-hippocampal injection of KA resulted in considerable neuronal loss in the hippocampal CA1 regions as assessed by immunohistochemical staining for NeuN. Administration of 80 mg/kg of MH reduced neuronal cell loss in the KA-treated hippocampus. When quantified and expressed as a percentage of preserved neurons in the area of interest of the ipsilateral compared with the contralateral CA1 controls, KA treatment significantly reduced the number of hippocampal neurons by 64% (Fig. 4B; ***p<0.001 vs. CON). However, treatment with MH (80 mg/kg) significantly protected the hippocampal neurons against KA-induced neurotoxicity (Fig. 4B; ##p<0.01 vs. KA-treated mice).
Extensive neuronal cell loss in the epileptic brain could be caused by excitotoxicity involved in apoptosis-mediated signaling pathways [40]. To examine whether morin treatment inhibits KA-induced apoptosis, the levels of c-caspase-3 and c-PARP-1, pro-apoptotic molecules, were measured by double-immunofluorescence staining and western blotting. As shown in Fig. 5A and B, double-immunofluorescence staining demonstrated that KA treatment significantly enhanced the expression of c-caspase-3 and c-PARP-1 in the hippocampal CA1 region, compared with controls, while the upregulation was diminished in mice treated with MH. Consistent with the immunofluorescence results, western blot analysis demonstrated that the increased levels of ccaspase-3 and c-PARP-1 in the KA-treated hippocampus (Fig. 5C and D; **p<0.01 and *p<0.05, respectively, vs. control mice) were significantly attenuated by treatment with MH, compared with KA alone (Fig. 5C and D; #p<0.05, respectively, vs. KA-treated mice).
The inflammatory events in a KA-induced rodent epilepsy model and in the hippocampus of patients with TLE are important to understand as they may relate to the pathological mechanisms involved in seizure-related neuronal cell death [7,41]. Thus, we examined whether morin treatment effects the expression of inflammatory mediators, such as TNF-α, IL-1β and iNOS, which may be produced by activated microglia [7], in the hippocampus of KA-treated mice. As shown in Fig. 6A, B, and C, immunofluorescence staining demonstrated that KA treatment resulted in microglial activation and an increase in TNF-α, IL-1β, and iNOS, in the hippocampus, while these increases in inflammatory mediators were inhibited by MH treatment. Consistent with these results, western blots also showed that MH treatment inhibited the increase in expression of TNF-α, IL-1β, and iNOS following the KA injection (Fig. 6D and E; #p<0.05 and ##p<0.01, respectively, vs. KA alone). Thus, these results suggest that morin protects hippocampal neurons by inhibiting the production of apoptotic signaling molecules and pro-inflammatory mediators.
DISCUSSION
Despite many new anti-epileptic drugs developed over the last two decades, anti-convulsant therapies are still limited and unable to control seizures in all patients [42]. To overcome these limitations, many studies have focused on the neuroprotective actions of flavonoids against brain injury induced by neurotoxins. The beneficial neuroprotective effects are thought to include the ability to suppress neuroinflammation and excitotoxicity with minimal toxic side effects compared with synthetic compounds [19,43]. Moreover, recent studies have reported anti-epileptic effects of some flavonoids in the KA-induced models of seizures [7,15,44], suggesting various natural flavonoids as alternative medicines for patients with epileptic seizures.
The formation of GCD, an abnormal widening of the GCL in the DG, was found in the surgical hippocampal specimens of patients with TLE [45]. The development of GCD in the hippocampus was influenced by abnormal activation of mTORC1 in rodent models of TLE.
Notably, inhibition of mTORC1 with rapamycin treatment markedly reduced the KA-induced GCD in the hippocampus [5,7,46], suggesting that activation of mTORC1 is an important risk factor in the progression of epilepsy.
Morin, a bioflavonoid constituent of many herbs and fruits, has been reported to prevent neurodegeneration and motor disability by reducing oxidative stress in a rat model of Huntington's disease [26]. Moreover, its beneficial effects, such as its anti-excitotoxicity, anti-apoptotic, and anti-inflammatory properties and its induction of neurotrophic factors were well described in previous studies [20,21,22,23,24,25,26,47]. However, it was largely unknown whether morin treatment affects the cytoarchitectural abnormalities associated with the progression of epilepsy and have neuroprotective effects against KA-induced neurotoxicity
Seizures induced by chemical convulsants, such as KA, can cause hippocampal neuronal death [48]. This neuronal death during chronic epilepsy may be due to apoptosis triggered by the activation of several signal transduction factors including caspase-3, PAPR-1, Bcl-2, and Bax [49,50]. This evidence suggests that controlling apoptosis may be important in protecting hippocampal neurons following epileptic seizures. Our results showed that MH treatment protected the hippocampal CA1 neuronal fibers and cell bodies against KA-induced excitotoxicity 2 days and 7 days postlesion (Fig. 4). Moreover, morin treatment prevented the cleavage of caspase-3 and PARP-1 following the KA injection (Fig. 5). Thus, these results suggested that MH treatment attenuated the apoptotic signal induced by KA treatment at early time points, resulting in protection from neuronal fiber degeneration. MH treatment also protected the hippocampal CA1 neurons against KA-induced neurotoxicity.
In addition to the increases in apoptotic signaling molecules, the increased levels of inflammatory mediators, including IL-1β, TNF-α, cyclooxygenase-2, and iNOS, may be involved in the neurotoxic events in the brain following epileptic seizures. The control of these inflammatory pathways may be one of the many potential strategies for the prevention and treatment of seizures [7,51,52,53]. In the present study, our results further demonstrated that morin treatment could inhibit microglial activation in the KA-treated hippocampus, resulting in a decrease in the levels of IL-1β, TNF-α, and iNOS (Fig. 6). These results suggest that the anti-apoptotic and anti-inflammatory effects of morin are associated with the protection of hippocampal neurons from KA-induced neurotoxicity.
In conclusion, we evaluated the beneficial effects of morin against KA-induced epileptic seizures
Figures

{kind=link}

{kind=link}

{kind=link}

{kind=link}

{kind=link}

{kind=link}
References
- Gourie-Devi M, Gururaj G, Satishchandra P, Subbakrishna DK. Prevalence of neurological disorders in Bangalore, India: a community-based study with a comparison between urban and rural areas. Neuroepidemiology 2004;23:261-268.

- Kobow K, Jeske I, Hildebrandt M, Hauke J, Hahnen E, Buslei R, Buchfelder M, Weigel D, Stefan H, Kasper B, Pauli E, Blümcke I. Increased reelin promoter methylation is associated with granule cell dispersion in human temporal lobe epilepsy. J Neuropathol Exp Neurol 2009;68:356-364.
- Houser CR. Granule cell dispersion in the dentate gyrus of humans with temporal lobe epilepsy. Brain Res 1990;535:195-204.
- Marucci G, Rubboli G, Giulioni M. Role of dentate gyrus alterations in mesial temporal sclerosis. Clin Neuropathol 2010;29:32-35.
- Na M, Liu Y, Shi C, Gao W, Ge H, Wang Y, Wang H, Long Y, Shen H, Shi C, Lin Z. Prognostic value of CA4/DG volumetry with 3T magnetic resonance imaging on postoperative outcome of epilepsy patients with dentate gyrus pathology. Epilepsy Res 2014;108:1315-1325.
- Shima A, Nitta N, Suzuki F, Laharie AM, Nozaki K, Depaulis A. Activation of mTOR signaling pathway is secondary to neuronal excitability in a mouse model of mesio-temporal lobe epilepsy. Eur J Neurosci 2015;41:976-988.
- Kim S, Jung UJ, Oh YS, Jeon MT, Kim HJ, Shin WH, Hong J, Kim SR. Beneficial effects of silibinin against kainic acid-induced neurotoxicity in the hippocampus
in vivo . Exp Neurobiol 2017;26:266-277.
- Park J, Jeong KH, Shin WH, Bae YS, Jung UJ, Kim SR. Naringenin ameliorates kainic acid-induced morphological alterations in the dentate gyrus in a mouse model of temporal lobe epilepsy. Neuroreport 2016;27:1182-1189.
- Zeng LH, Rensing NR, Wong M. The mammalian target of rapamycin signaling pathway mediates epileptogenesis in a model of temporal lobe epilepsy. J Neurosci 2009;29:6964-6972.
- Baram TZ, Eghbal-Ahmadi M, Bender RA. Is neuronal death required for seizure-induced epileptogenesis in the immature brain?. Prog Brain Res 2002;135:365-375.
- Vezzani A, French J, Bartfai T, Baram TZ. The role of inflammation in epilepsy. Nat Rev Neurol 2011;7:31-40.
- Samland H, Huitron-Resendiz S, Masliah E, Criado J, Henriksen SJ, Campbell IL. Profound increase in sensitivity to glutamatergic- but not cholinergic agonist-induced seizures in transgenic mice with astrocyte production of IL-6. J Neurosci Res 2003;73:176-187.
- Henshall DC. Apoptosis signalling pathways in seizure-induced neuronal death and epilepsy. Biochem Soc Trans 2007;35:421-423.
- Rocha LL, Lopez-Meraz ML, Niquet J, Wasterlain CG. Do single seizures cause neuronal death in the human hippocampus?. Epilepsy Curr 2007;7:77-81.
- Louis ED, Williamson PD, Darcey TM. Experimental models of chronic focal epilepsy: a critical review of four models. Yale J Biol Med 1987;60:255-272.
- Lee J, Lim E, Kim Y, Li E, Park S. Ghrelin attenuates kainic acid-induced neuronal cell death in the mouse hippocampus. J Endocrinol 2010;205:263-270.
- Wang Q, Yu S, Simonyi A, Sun GY, Sun AY. Kainic acid-mediated excitotoxicity as a model for neurodegeneration. Mol Neurobiol 2005;31:3-16.
- Mohd Sairazi NS, Sirajudeen KN, Asari MA, Muzaimi M, Mummedy S, Sulaiman SA. Kainic acid-induced excitotoxicity experimental model: protective merits of natural products and plant extracts. Evid Based Complement Alternat Med 2015;2015:972623.
- Greenwell M, Rahman PK. Medicinal plants: their use in anticancer treatment. Int J Pharm Sci Res 2015;6:4103-4112.
- Manna SK, Aggarwal RS, Sethi G, Aggarwal BB, Ramesh GT. Morin (3,5,7,2′,4′-pentahydroxyflavone) abolishes nuclear factor-kappaB activation induced by various carcinogens and inflammatory stimuli, leading to suppression of nuclear factor-kappaB-regulated gene expression and upregulation of apoptosis. Clin Cancer Res 2007;13:2290-2297.
- Gálvez J, Coelho G, Crespo ME, Cruz T, Rodríguez-Cabezas ME, Concha A, Gonzalez M, Zarzuelo A. Intestinal anti-inflammatory activity of morin on chronic experimental colitis in the rat. Aliment Pharmacol Ther 2001;15:2027-2039.
- Yu Z, Fong WP, Cheng CH. The dual actions of morin (3,5,7,2′,4′-pentahydroxyflavone) as a hypouricemic agent: uricosuric effect and xanthine oxidase inhibitory activity. J Pharmacol Exp Ther 2006;316:169-175.
- Lian TW, Wang L, Lo YH, Huang IJ, Wu MJ. Fisetin, morin and myricetin attenuate CD36 expression and oxLDL uptake in U937-derived macrophages. Biochim Biophys Acta 2008;1781:601-609.
- Caillet S, Yu HL, Lessard S, Lamoureux G, Ajdukovic D, Lacroix M. Fenton reaction applied for screening natural antioxidants. Food Chem 2007;100:542-552.
- Gottlieb M, Leal-Campanario R, Campos-Esparza MR, Sánchez-Gómez MV, Alberdi E, Arranz A, Delgado-García JM, Gruart A, Matute C. Neuroprotection by two polyphenols following excitotoxicity and experimental ischemia. Neurobiol Dis 2006;23:374-386.
- Rao Ac, Shende V. Morin hydrate prevents neurodegeneration in 3-nitropropionic acid induced Huntington's disease. Am J Pharmtech Res 2015;5:227-241.
- Phelan KD, Shwe UT, Williams DK, Greenfield LJ, Zheng F. Pilocarpine-induced status epilepticus in mice: a comparison of spectral analysis of electroencephalogram and behavioral grading using the Racine scale. Epilepsy Res 2015;117:90-96.
- Racine RJ. Modification of seizure activity by electrical stimulation. II. Motor seizure. Electroencephalogr Clin Neurophysiol 1972;32:281-294.
- Jeong KH, Jung UJ, Kim SR. Naringin attenuates autophagic stress and neuroinflammation in kainic acid-treated hippocampus
in vivo . Evid Based Complement Alternat Med 2015;2015:354326.
- Butler CR, Boychuk JA, Smith BN. Effects of rapamycin treatment on neurogenesis and synaptic reorganization in the dentate gyrus after controlled cortical impact injury in mice. Front Syst Neurosci 2015;9:163.
- Hunt RF, Scheff SW, Smith BN. Posttraumatic epilepsy after controlled cortical impact injury in mice. Exp Neurol 2009;215:243-252.
- Shibley H, Smith BN. Pilocarpine-induced status epilepticus results in mossy fiber sprouting and spontaneous seizures in C57BL/6 and CD-1 mice. Epilepsy Res 2002;49:109-120.
- Jeon MT, Nam JH, Shin WH, Leem E, Jeong KH, Jung UJ, Bae YS, Jin YH, Kholodilov N, Burke RE, Lee SG, Jin BK, Kim SR.
In vivo AAV1 transduction with hRheb(S16H) protects hippocampal neurons by BDNF production. Mol Ther 2015;23:445-455.
- Kim HD, Jeong KH, Jung UJ, Kim SR. Myricitrin ameliorates 6-hydroxydopamine-induced dopaminergic neuronal loss in the substantia nigra of mouse brain. J Med Food 2016;19:374-382.
- Macias M, Blazejczyk M, Kazmierska P, Caban B, Skalecka A, Tarkowski B, Rodo A, Konopacki J, Jaworski J. Spatiotemporal characterization of mTOR kinase activity following kainic acid induced status epilepticus and analysis of rat brain response to chronic rapamycin treatment. PLoS One 2013;8:e64455.
- McDaniel SS, Wong M. Therapeutic role of mammalian target of rapamycin (mTOR) inhibition in preventing epileptogenesis. Neurosci Lett 2011;497:231-239.
- Meng XF, Yu JT, Song JH, Chi S, Tan L. Role of the mTOR signaling pathway in epilepsy. J Neurol Sci 2013;332:4-15.
- Wong M. Mammalian target of rapamycin (mTOR) inhibition as a potential antiepileptogenic therapy: from tuberous sclerosis to common acquired epilepsies. Epilepsia 2010;51:27-36.
- Bertoglio D, Amhaoul H, Van Eetveldt A, Houbrechts R, Van De Vijver S, Ali I, Dedeurwaerdere S. Kainic acid-induced post-status epilepticus models of temporal lobe epilepsy with diverging seizure phenotype and neuropathology. Front Neurol 2017;8:588.
- Dingledine R, Varvel NH, Dudek FE. When and how do seizures kill neurons, and is cell death relevant to epileptogenesis?. Adv Exp Med Biol 2014;813:109-122.
- Ravizza T, Balosso S, Vezzani A. Inflammation and prevention of epileptogenesis. Neurosci Lett 2011;497:223-230.
- Lasoń W, Leśkiewicz M. Effect of plant polyphenols on seizures - animal studies. J Epileptology 2013;21:79-87.
- Bagli E, Goussia A, Moschos MM, Agnantis N, Kitsos G. Natural compounds and neuroprotection: mechanisms of action and novel delivery systems. In Vivo 2016;30:535-547.
- Sahranavard S, Ghafari S, Mosaddegh M. Medicinal plants used in Iranian traditional medicine to treat epilepsy. Seizure 2014;23:328-332.
- Bae EK, Jung KH, Chu K, Lee ST, Kim JH, Park KI, Kim M, Chung CK, Lee SK, Roh JK. Neuropathologic and clinical features of human medial temporal lobe epilepsy. J Clin Neurol 2010;6:73-80.
- Jang H, Jeong KH, Kim SR. Naringin attenuates granule cell dispersion in the dentate gyrus in a mouse model of temporal lobe epilepsy. Epilepsy Res 2016;123:6-10.
- Zhang ZT, Cao XB, Xiong N, Wang HC, Huang JS, Sun SG, Wang T. Morin exerts neuroprotective actions in Parkinson disease models
in vitro andin vivo . Acta Pharmacol Sin 2010;31:900-906.
- Pitkänen A, Kharatishvili I, Karhunen H, Lukasiuk K, Immonen R, Nairismägi J, Gröhn O, Nissinen J. Epileptogenesis in experimental models. Epilepsia 2007;48:13-20.
- Tokuhara D, Sakuma S, Hattori H, Matsuoka O, Yamano T. Kainic acid dose affects delayed cell death mechanism after status epilepticus. Brain Dev 2007;29:2-8.
- Niquet J, Auvin S, Archie M, Seo DW, Allen S, Sankar R, Wasterlain CG. Status epilepticus triggers caspase-3 activation and necrosis in the immature rat brain. Epilepsia 2007;48:1203-1206.
- Vezzani A, Baram TZ. New roles for interleukin-1 Beta in the mechanisms of epilepsy. Epilepsy Curr 2007;7:45-50.
- Wilcox KS, Vezzani A. Does brain inflammation mediate pathological outcomes in epilepsy?. Adv Exp Med Biol 2014;813:169-183.
- Webster KM, Sun M, Crack P, O'Brien TJ, Shultz SR, Semple BD. Inflammation in epileptogenesis after traumatic brain injury. J Neuroinflammation 2017;14:10.