Articles

Article Tools
 View Full Text View Full Text |
 Abstract Abstract |
 Article as PDF Article as PDF |
 Print this Article Print this Article |
 Pubmed Pubmed |
 PMC PMC |
 PubReader PubReader |
 Export to Citation Export to Citation |
 Email Alerts Email Alerts |
 Open Access Open Access |
Share this article on :



Stats or Metrics
Article
Original Article
Exp Neurobiol 2019; 28(5): 612-627
Published online October 31, 2019
https://doi.org/10.5607/en.2019.28.5.612
© The Korean Society for Brain and Neural Sciences
Transduced Tat-aldose Reductase Protects Hippocampal Neuronal Cells against Oxidative Stress-induced Damage
Su Bin Cho1†, Won Sik Eum1†, Min Jea Shin1†, Hyun Jung Kwon2, Jung Hwan Park1, Yeon Joo Choi1, Jinseu Park1, Kyu Hyung Han1, Ju Hyeon Kang3, Duk-Soo Kim3, Sung-Woo Cho4, Dae Won Kim2* and Soo Young Choi1*
1Department of Biomedical Science and Research Institute of Bioscience and Biotechnology, Hallym University, Chuncheon 24252, 2Department of Biochemistry and Molecular Biology, Research Institute of Oral Sciences, College of Dentistry, Gangneung-Wonju National University, Gangneung 25457, 3Department of Anatomy, College of Medicine, Soonchunhyang University, Cheonan 31538, 4Department of Biochemistry and Molecular Biology, University of Ulsan College of Medicine, Seoul 05505, Korea
Correspondence to: *To whom correspondence should be addressed.
Soo Young Choi, TEL: 82-33-248-2112, FAX: 82-33-248-3202
e-mail: sychoi@hallym.ac.kr
Dae Won Kim, TEL: 82-33-640-2229, FAX: 82-33-642-6410
e-mail: kimdw@gwnu.ac.kr
†These authors contributed equally to this work.
This is an Open Access article distributed under the terms of the Creative Commons Attribution Non-Commercial License(http://creativecommons.org/licenses/by-nc/4.0) which permits unrestricted non-commercial use, distribution, andreproduction in any medium, provided the original work is properly cited.
Abstract
Aldose reductase (AR) protein, a member of the NADPH-dependent aldo-keto reductase family, reduces a wide range of aldehydes and enhances cell survival by inhibition of oxidative stress. Oxidative stress is known as one of the major pathological factor in ischemia. Since the precise function of AR protein in ischemic injury is fully unclear, we examined the function of AR protein in hippocampal neuronal (HT-22) cells and in an animal model of ischemia in this study. Cell permeable Tat-AR protein was produced by fusion of protein transduction domain in Tat for delivery into the cells. Tat-AR protein transduced into HT-22 cells and significantly inhibited cell death and regulated the mitogen-activate protein kinases (MAPKs), Bcl-2, Bax, and Caspase-3 under oxidative stress condition. In an ischemic animal model, Tat-AR protein transduced into the brain tissues through the blood-brain barrier (BBB) and drastically decreased neuronal cell death in hippocampal CA1 region. These results indicate that transduced Tat-AR protein has protective effects against oxidative stress-induced neuronal cell death
Graphical Abstract
Keywords: Tat-AR, Oxidative stress, Ischemia, MAPKs, Cytotoxicity, Protein therapy
INTRODUCTION
Cerebral ischemia is a neuronal disease that occurs when the blockage of blood flow to the brain causes reperfusion injury and insufficient amount of oxygen induced by oxidative stress leads to neuronal cell death [1, 2]. It is well known that the high levels of oxidative stress critically increase neuronal cell death, whereas inhibition of oxidative stress by overexpression of antioxidant proteins reduced neuronal cell death [3]. Many studies have shown that high levels of oxidative stress destroy intracellular macromolecules including DNA and proteins and finally leads to cell death [4, 5]. Conversely, natural products and proteins such as curcumin and caveolin-1/-3, which have antioxidant functions, have been shown to inhibit cell death including ischemic insults via the inhibition of oxidative stress [6, 7].
Aldose reductase (AR; EC 1.1.1.21), a member of the NADPH-dependent aldo-keto reductase family, reduces aldehydes detoxification during lipid peroxidation and several studies have demonstrated that AR plays protective roles as an antioxidant protein against reactive oxygen species (ROS) [8–10]. Thus this protein detoxifies ROS induced lipid aldehyde materials and protects against cell death via the inhibition of oxidative stress in a variety of cells such as SH-SY5Y cells [10]. Other studies also showed that AR protein enhanced cell survival by inhibiting cell toxicities in smooth muscle and lens epithelial cells [11].
Protein transduction domains (PTDs) carries not only proteins and peptides but also antisense, plasmids, microbeads, liposomes, and other molecules into cells without any special receptor [12]. Since PTD-fused proteins can pass through the membrane and enter into the inner part of cell, cell permeable PTD like Tat-peptide can be useful tools in protein therapy when it fused with target protein [13, 14]. We have demonstrated that various PTD fusion proteins showed protective effect against cell damage
In this study, we fused AR with Tat PTD to transduce into cells and examined whether this Tat-AR fusion protein protects against oxidative stress-induced hippocampal HT-22 cell death and in an ischemic animal models.
MATERIALS AND METHODS
HT-22 cell culture and materials
Mouse hippocampal HT-22 cells were grown in Dulbecco’s modified Eagle’s medium (DMEM) containing 10% fetal bovine serum and antibiotics (100 μg/ml streptomycin, 100 μg /ml penicillin) at 37°C in a humidity chamber with 5% CO2 and 95% air.
Ni2+-nitrilotriacetic acid Sepharose Superflow was purchased from Qiagen (Valencia, CA, USA). PD-10 columns were purchased from Amersham (Braunschweig, Germany). The indicated primary and β-actin antibodies were obtained from Cell Signaling Technology (Beverly, MA, USA) and Santa Cruz Biotechnology (Santa Cruz, CA, USA). An enzyme-linked immunosorbent assay (ELISA) kit for hexa histidine was obtained from Cloud-Clone Corp. (Houstern, TX, USA). Unless otherwise stated, all other agents were of the highest grade available.
Purification and transduction of Tat-AR protein into HT-22 cells
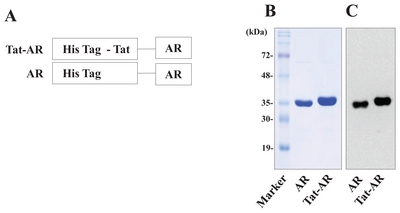
Preparation of the Tat expression vector has been described in a previous study [15]. Human AR was amplified by PCR with two primers. The sense primer 5′-CTCGAGGCAAGCCGTCTCCT-3′ contained an
The antisense primer 5′-GGATCCTCAAAACTCTTCATGGAAGG-3′ contained a
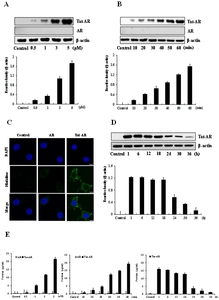
To examine whether Tat-AR protein transduced efficiently in a time and concentration dependent, HT-22 cells were exposed to different concentrations (0.5~5 μM) of Tat-AR and AR protein for 1 h. HT-22 cells were exposed 5 μM of Tat-AR and AR protein for various time periods (10~60 min). Cells were then washed with trypsin-EDTA and washed twice with PBS. The amounts of transduced proteins were measured by Western blotting. We also determined the intracellular stability of Tat-AR protein by culturing the cells (1~36 h) after transduction. Then transduced levels were measured by Western blotting using an anti-histidine antibody.
Western blot analysis
Equal amounts of proteins were loaded into 12% SDS-PAGE and electrotransferred to a polyvinylidene difluoride (PVDF) membrane. The membrane was blocked with TBS-T (25 mM Tris-HCl, 140 mM NaCl, 0.1% Tween 20, pH 7.5) buffer containing 5% nonfat dry milk for 1 h. After being washed with TBST, the membrane was incubated with the indicated primary and appropriate secondary antibodies recommended by the manufacturer. Then the membranes were washed with TBST buffer three times and the protein bands were identified using chemiluminescent reagents as recommended by the manufacturer (Amersham, Franklin Lakes, NJ, USA) [16].
Measurement of transduced Tat-AR protein levels
HT-22 cells (1×106) were pretreated with Tat-AR proteins and AR (0.5~5 μM for 1 h or 5 μM for 10~60 min). Cells were then washed with PBS and treated with trypsin-EDTA. Transduced Tat-AR protein levels were analyzed using an ELISA kit for hexa histidine (Cloud-Clone Corp.) according to the manufacture’s instruction.
Confocal fluorescence microscopy analysis
To determine the intracellular distribution of transduced Tat-AR protein in HT-22 cells, we performed confocal fluorescence microscopy as described previously [16, 17]. HT-22 cells were placed on coverslips and treated with 5 μM of Tat-AR protein for 1 h. The cells were washed with PBS twice and fixed with 4% paraformaldehyde for 5 min. The cells were treated in PBS containing 3% bovine serum albumin and 0.1% Triton X-100 (PBS-BT) at room temperature for 30 min and washed with PBS-BT. The histidine primary antibody was diluted 1:1500 and incubated at room temperature for 3 h. The Alexa Fluor 488-conjugated secondary antibody (Invitrogen, Carlsbad, CA, USA) was diluted 1:1500 and incubated in the dark for 1 h. Nuclei were stained with 1 μg/ml DAPI (Roche Applied Science, Mannheim, Germany) for 2 min. Then stained cells were analyzed by confocal fluorescence microscopy using a confocal laser-scanning system (Bio-Rad MRC-1024ES, 4BIOROD, CA, USA).
Cell viability assay
Cell viability was measured using a 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay as described previously [19, 24]. HT-22 cells were seeded on a 96-well plate and treated with Tat-AR or AR protein for 1 h. Then the cells were incubated with 1 mM hydrogen peroxide (H2O2) for 8 h. The absorbance was determined at 540 nm using a microplate reader (Infinite 200 nanoquant, TECAN, Switzerland) and cell viability was defined as the percentage of untreated control cells.
DNA fragmentation staining
To examine whether transduced Tat-AR proteins protect against H2O2-induced DNA damage in cells, HT-22 cells were pretreated with 5 μM Tat-AR protein for 1 h and exposed to 1 mM H2O2 for 6 h. Terminal deoxynucleotidyl transferase-mediated biotinylated dUTP nick end labeling (TUNEL) staining was performed using a Cell Death Detection kit (Roche Applied Science). Each fluorescent image was obtained using a fluorescence microscope (Nikon eclipse 80i, Tokyo, Japan). Fluorescence positive cells were counted under a phage-contrast microscopy (× 200 magnification) [16, 17].
Experimental animals and treatment
Male gerbils (65~75 g, 6 months) obtained from the Hallym University Experimental Animal Center, were housed at a temperature of 23ºC, with humidity of 60%, and exposed to 12 hour periods of light and dark with free access to food and water. All experimental procedures involving animals and their care conformed to the Guide for the Care and Use of Laboratory Animals of the National Veterinary Research & Quarantine Service of Korea and were approved by the Institutional Animal Care and Use Committee of Soonchunhyang University [SCH16-0009].
The transient forebrain ischemia model was performed as described previously [16, 17]. Briefly, the animals were anesthetized, common carotid arteries were isolated, freed of nerve fibers, and occluded with non-traumatic aneurysm clips. Complete interruption of blood flow was confirmed by observing the retinal artery using an ophthalmoscope. After 5 min occlusion, the aneurysm clips were removed. The restoration of blood flow (reperfusion) was observed directly under the ophthalmoscope.
To explore the protective effects of Tat-AR protein against ischemic damage, the animals were divided into 4 groups (each n=10): control sham group, vehicle-treated group, AR-treated group, and Tat-AR-treated groups. The AR and Tat-AR proteins (2 mg/kg) were administered intraperitoneally 30 min before ischemia-reperfusion.
Immunohistochemistry
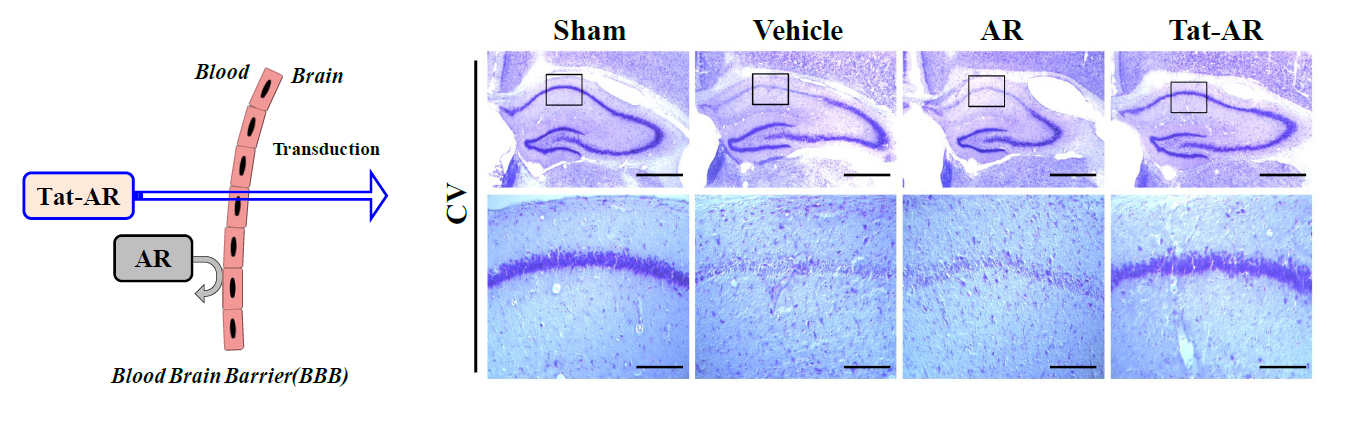
Immunohistochemistry was performed as described in previous studies [16–18]. Brain tissue samples were obtained at 7 days after ischemia-reperfusion. To examine the protective effects of transduced Tat-AR protein against ischemic damage, the sections were incubated in 10% normal goat serum in PBS for 30 min and the sections were stained with a histidine antibody, Cresyl violet (CV), Fluoro-Jade B (FJB), ionized calcium-binding adapter molecule 1 (Iba-1) and neuronal nuclei (NeuN).
The positive neuronal cell number and intensity of immunoreactivity were calculated using an image analyzing system equipped with a computer based CCD camera (software: Optimas 6.5, CyberMetrics, USA). The staining intensity of the immunoreactive structures was evaluated as the relative optical density (ROD). A ratio of the ROD was calibrated as % [16, 17].
Statistical analysis
Data are expressed as the mean±SEM of three experiments. Differences between groups were analyzed by ANOVA followed by a Bonferroni’s post-hoc test. Statistical significance was considered at p<0.05.
RESULTS
Purification and transduction of Tat-AR protein into HT-22 cells
A human AR gene was fused with a Tat PTD to produce cell-permeable Tat-AR protein. Also, we constructed an AR protein expression vector as a control (Fig. 1A). As shown in Fig. 1B and 1C, Tat-AR and AR proteins were purified and confirmed by SDS-PAGE and Western blotting. Purified Tat-AR and AR proteins showed to the expected molecular weights of approximately 37 and 36 kDa, respectively.
To investigate whether Tat-AR and AR protein possesses the capacity to transduce into HT-22 cells, the cells were treated with various concentrations of Tat-AR proteins (0.5~5 μM) protein for 1 h or various time periods (10~60 min) of Tat-AR proteins (5 μM). Then, cells were washed with PBS and cell lysates were analyzed by Western blotting. Tat-AR proteins were detected in the cell lysates from transduced HT-22 cells both a concentration- and time-dependently (Fig. 2A and 2B). Also, we determined the distribution of transduced proteins in HT-22 cells using Alexa Fluor 488 and DAPI immunostaining. As expected, the transduced Tat-AR proteins were detected in the cytoplasm and nucleus of the cells. In contrast, AR protein was not transduced under the same conditions (Fig. 2C). Furthermore, we assessed the stability of transduced Tat-AR protein. HT-22 cells were incubated with different time periods (1~36 h) after transduction of Tat-AR protein to persist in the cells. Tat-AR protein was observed in the cells up to 36 h (Fig. 2D). We also examined the transduced Tat-AR protein levels using an ELISA kit. As shown in Fig. 2E, transduced Tat-AR protein levels were increased concentration- and time-dependently.
Effects of Tat-AR protein against H2O2-induced HT-22 cell damages
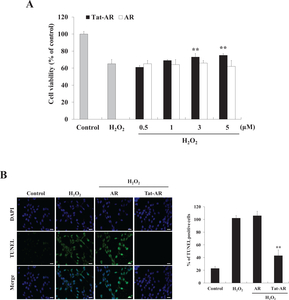
The protective effects of Tat-AR protein against H2O2-induced HT-22 cell damages were determined, as shown in Fig. 3. To examine the effect of Tat-AR protein on cell viability, we performed an MTT assay. Cell viability was about 60% in the cells treated with only H2O2 (1 mM, 8 h), whereas Tat-AR protein increased cell viability in an accordance with Tat-AR concentration up to 75% (Fig. 3A).
To determine whether Tat-AR protein inhibits DNA fragmentation, TUNEL staining was performed. DNA fragmentation significantly increased in the cells treated with only H2O2 (1 mM, 3 h) treatment cells. Tat-AR protein markedly inhibited DNA fragmentation (Fig. 3B).
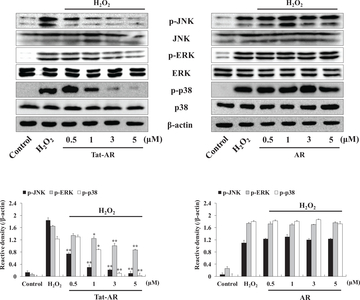
Effects of Tat-AR proteins on H2O2-induced signaling pathways in HT-22 cells
The mitogen-activated protein kinases (MAPKs) signaling pathways, such as extracellular signal regulating kinase 1/2 (ERK1/2 or p44/42), c-Jun N-terminal kinase (JNK), and p38, are highly associated with ROS and finally lead to cell death [25]. We found that Tat-AR protein markedly and concentration-dependently reduced the expression of MAPKs phosphorylation in cells treated with H2O2 (1 mM). However, AR protein displayed no change in expression levels compared to the cells treated with H2O2 alone (Fig. 4).
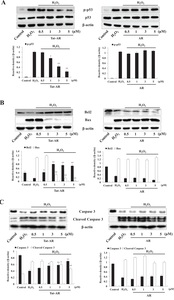
Next, we examined whether Tat-AR protein inhibits H2O2-induced apoptosis because H2O2 is known to induce apoptosis. Phosphorylation p53 expression levels cause apoptosis [26, 27]. As shown in Fig. 5A, Tat-AR protein markedly inhibited the phosphorylated p53 expression concentration-dependently compared to the only H2O2 treated cells. Bax, Bcl-2, and Caspase-3 protein expression are associated with the oxidative stress induced apoptotic processes [28–30]. We showed that Tat-AR protein concentration-dependently elevated the Bcl-2 expression in the H2O2 treated cells, whereas the expression of Bax showed the opposite effect compared to the Bcl-2 (Fig. 5B). Further, we showed that Caspase-3 expression reduced in the H2O2 only treated cells. However, Tat-AR protein drastically increased the Caspase-3 expression and cleaved Caspase-3 expression showed the opposite effect compared to Caspase-3 (Fig. 5C). However, AR protein did not show the same changes of expression of apoptotic related proteins under the same experimental conditions.
Protective effect of Tat-AR protein on ischemic injury
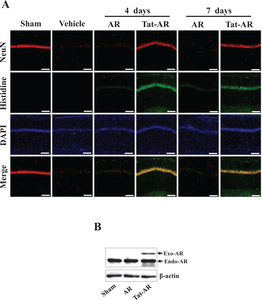
To investigate the protective effects of transduced Tat-AR protein on ischemic injury in an animal model, we performed immunohistochemistry using a histidine antibody and NeuN staining. NeuN is known as a marker for neurons generally used to detect neurons. As shown in Fig. 6A, we observed that Tat-AR protein transduced into the hippocampal CA1 region, crossing the blood-brain barrier (BBB), where it markedly protected against neuronal cell death compared to the vehicle- and AR protein treated groups. We also showed that transduced Tat-AR protein significantly increased hippocampal neuronal cell survival. However, the AR protein treated group showed a similar pattern compared with vehicle treated group. To investigate the endogenous levels of AR protein in gerbil brain, gerbils treated with Tat-AR protein were sacrificed and obtained brain tissue. Then, endogenous AR protein levels were confirmed by Western blotting with AR antibody. As shown in Fig. 6B, transduced Tat-AR protein doesn’t affect the level of endogenous AR protein significantly.
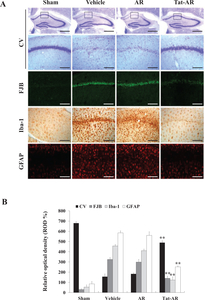
Furthermore, we examined whether transduced Tat-AR protein inhibits neuronal cell injury and activation of microglia and astrocytes using FJB, Iba-1, and GFAP staining (Fig. 7). In the vehicle-and AR protein-treated groups, FJB, Iba-1, and GFAP fluorescence signals were intensively detected in the hippocampal CA1 region. In contrast, intensively fluorescence signals were markedly reduced in the Tat-AR treated group. These results indicate that Tat-AR protein transduced into hippocampal CA1 region, traversing the BBB, and protected against neuronal cell damage resulting from ischemic injury by decreasing microglia and astrocyte activation.
DISCUSSION
The generation of ROS induced by oxidative stress is involved in the pathogenesis of neurodegenerative disorders, cancers, and inflammatory diseases. Since excessive oxidative stress induces serious cell damage and finally leads to cell death, the inhibition of excessive oxidative stress may prevent various disorders [31–33]. The role of AR protein in a variety of disorders has been widely investigated. Several studies have shown that expression of AR protein protects against ROS formation and plays an important role as an antioxidant in neuronal cell [9–11]. On the other hand, some studies have demonstrated that inhibition of AR dramatically prevents production of LPS-induced cytokines, and inflammatory mediator proteins in Raw 264.7 cells, suggesting that inhibitors of AR could be used for therapeutic agent in inflammation [34]. Thus, the AR protein shows contradictory effects depending on cell type or disease. Therefore, we examined whether AR protein has a protective effect against oxidative stress-induced HT-22 cell death and in an ischemic injury animal model.
Many studies have demonstrated that PTD fusion proteins transduce into cells and tissues suggesting that PTD fusion proteins can be attractive therapeutic tools for various diseases [12–14, 35]. Tat, human immunodeficiency virus transactivator of transcript, is identified as the 11 amino acid sequence YGRKKRRQRRR and Tat fusion protein has ability to deliver a variety of proteins crossing the cell membranes and the BBB. Thus, transduction of Tat PTD fusion protein can represent a novel strategy for treating a majority of neuronal disorders [36, 37]. In previous studies, we reported that transduced Tat fused proteins have protective effects against oxidative stress induced cell deaths and in animal models of various diseases [15–22].
It is well known that excessive cellular ROS generation leads to cell death and macromolecules damage including DNA [4, 5]. Therefore we investigated the cell viability and TUNEL staining assay to show the protective effect of Tat-AR against ROS. The data showed that Tat-AR protein inhibits cell death and DNA fragmentation induced by oxidative stress. Several studies have shown that AR protein inhibits ROS toxicity. Overexpression of AR protein increased human lens epithelial cell survival in aldehyde-induced toxic condition and inhibited UVB-induced cell death and intracellular ROS generation in HaCaT cell. Thus, they suggested that AR proteins inhibit the oxidative stress by sequestering ROS [11, 38]. Our results showed same patterns as those reports suggesting Tat-AR protein protected H2O2-induced cell death and ROS generation in HT-22 cells.
Next, we investigated the effect of Tat-AR protein against oxidative stress-induced MAPKs signaling pathways. We showed that transduced Tat-AR protein inhibited the activation of MAPKs in the HT-22 cells. Increased ROS generation is known highly associated with cell signaling pathways by stimulation of redox-sensitive transcription factors and MAPKs (JNK, ERK1/2 and p38), which are known as a superfamily of serine/threonine kinases [25]. In smooth muscle cells (SMC), overexpression of AR protein is involved in the methylglyoxal (MG)- and hydrogen peroxide-induced p38 and ERK signaling pathways [39]. Another study also showed that transfection of AR gene reduced the ultraviolet-B (UVB)-mediated activation of MAPKs (p38 and JNK) in HaCaT cells while the ERK was not affected in HaCaT cells [38]. Those studies showed coincidence with our data suggesting that AR protein inhibits the activation of MAPKs signaling pathways.
In a previous studies have shown that excessive ROS generation leads to DNA damage and cell death by mediating apoptotic signaling pathways [40, 41]. Thus, we examined the effect of Tat-AR protein on apoptotic-related protein expression including p53, Bax, Bcl-2, and Caspase-3 under oxidative stress condition. Our data showed that transduced Tat-AR protein reduced the phosphorylation of p53, Bax, and cleaved Caspase-3 expression, while Bcl-2 expression were significantly increased in the oxidative stress-induced HT-22 cells. Kang et al (2011) demonstrated that AR protein prevents activation of p53 expression levels in keratinocytes which are exposed to UVB. This protein also suppressed the activation of Caspase-3 expression levels and markedly attenuated Bax and Bcl-2 expression in UV-B treated cells, suggesting that overexpression of AR protein inhibits UVB-induced apoptotic cell death via the regulation of apoptotic protein expression [38]. Although further studies are necessary to clarify the precise role of AR in MAPK and apoptotic signaling pathways, those results suggested that AR protein plays a detoxification role and increases cell survival via regulation of MAPKs and apoptotic signaling pathways under oxidative stress conditions.
Oxidative stress-induced ROS plays a crucial role in brain injury following ischemia-reperfusion [42–45]. It has been reported that oxidative stress induced accumulation of 4-hydroxy-2-nonenal (4-HNE) and malondialdehyde (MDA) during ischemic injury [46, 47]. Thus, these authors suggest that the balance between antioxidant and oxidative stress in organism might be the best approach for protection against ischemic damage. As
In a previous study, Cho et al (2008) have demonstrated that cell permeable PTD-GFP fusion protein transduced into ischemic hippocampal neurons and transduced PTD-GFP fusion protein levels were persisted over 4 days in ischemic animal model [55]. Usually the stability of most transduced proteins in the cells is about 24~72 hours depending on the cell types or proteins, however the stability of transduced proteins in brain tissues is about 4~7 days [17, 55–58]. The differences of stability between
It is known that Iba-1 and GFAP are generally expressed in microglia and astrocytes of the intact brain and its expression under ischemic injury is increased. Thus, the enhanced expressional patterns of Iba-1 and GFAP in the brain are considered as markers of microglia and astrocyte activities under ischemic injury [59, 60]. In previous studies we have demonstrated that the changes of glia’s activation after administration of various proteins were observed in ischemic animal model [17, 18, 57].
Other studies have shown that activation of microglia protects neuronal cell death by inhibition of inflammation at early stage of neuronal diseases [61–63]. Microglia are the principal immune cells in the central nervous system and microglial activation has dual effects (pro-inflammatory; M1-like or anti-inflammatory/protective; M2-like) by the release of a number of inflammatory mediators in neuronal diseases including ischemic injury [64, 65]. In the mild activated microglial cells contributes to restore the tissue homeostasis by clearing pathogens, necrotic cells and suppressing the inflammation, and facilitating the brain repair [66, 67]. However, over-activated microglial cells may exacerbate tissue damage and neuronal cell death by excessive production of neurotoxic substances including cytokines, nitric oxide (NO), and ROS [68, 69]. Thus, since microglial activation has the dual roles like promoting beneficial and detrimental effects on neurons, regulation of balance between beneficial and detrimental effects of microglial responses may be important in ischemic injury.
NF-κB and MAPK signaling pathways considered to be one of critical regulators and plays a major role in the inflammatory responses by controlling the activation of microglia [70, 71]. In the inactivated state of microglia, NF-κB is present in the cytoplasm in inactive form. However, NF-κB translocates into the nucleus when microglia are activated and regulates the gene expression of pro-inflammatory mediators. Some studies have shown that the expression of pro-inflammatory cytokines increases within hours and elevates at several weeks later in ischemic stroke [72–74]. MAPK is a crucial signaling pathway that mediates inflammation and participates in cytokine control [75]. Dong et al. (2019) have shown that oxymatrine (OMT) alleviates neuronal damage and improves hippocampal neuronal states by inhibition of microglia activation and MAPK signal pathways activation in rat brain tissues and primary microglia cells [76]. Furthermore, there are many evidences suggest that inhibition of microglia activation is important for neuroprotection in ischemic injury [77–80], therefore how to reduce of pro-inflammatory mediators from activated microglia is one of factors to find out the therapeutic molecules against ischemic injury [81–86].
In this study, after treatment of Tat-AR protein to ischemic animal model, the patterns of the changes of microglia and astrocyte activation showed coincidence with the protection of the hippocampal neuronal cells obtained by CV and FJB staining experiments. These our results indicate that transduced Tat-AR protein play an important role for hippocampal neuronal cell survival as an antioxidant function. However, further studies are necessary to understand the precise protective molecular mechanisms and functions of AR protein during ischemic injury.
In summary, we demonstrated that Tat-AR protein transduced into HT-22 cells and significantly protected cell death caused by oxidative stress via inhibition of DNA fragmentation and regulation of MAPK and apoptotic signaling pathways. In addition, Tat-AR protein transduced into hippocampal neuronal cell and prevents cell death in an ischemic animal model. Thus, we suggested that Tat-AR protein can be a potential therapeutic protein agent for ischemic injury.
ACKNOWLEDGEMENTS
This work was supported by the National Research Foundation of Korea (NRF) grant funded the Korea government (MIST) (NRF-2018R1A2B6001941) and in part by the Bio & Medical Technology Development Program of the National Research Foundation (NRF) funded by the Korean government (MSIT) (NRF-2018M3A9C8023568). Also, this research was supported by Basic Science Research Program through the National Foundation of Korea funded by the Ministry of Education (2019R1A6A1A11036849).
Figures
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
References
- Chan PH (2001) Reactive oxygen radicals in signaling and damage in the ischemic brain. J Cereb Blood Flow Metab 21:2-14.


- Chan PH (1996) Role of oxidants in ischemic brain damage. Stroke 27:1124-1129.
- Niizuma K, Endo H, Chan PH (2009) Oxidative stress and mitochondrial dysfunction as determinants of ischemic neuronal death and survival. J Neurochem 109 Suppl 1:133-138.

- Kujoth GC, Hiona A, Pugh TD, Someya S, Panzer K, Wohlgemuth SE, Hofer T, Seo AY, Sullivan R, Jobling WA, Morrow JD, Van Remmen H, Sedivy JM, Yamasoba T, Tanokura M, Weindruch R, Leeuwenburgh C, Prolla TA (2005) Mitochondrial DNA mutations, oxidative stress, and apoptosis in mammalian aging. Science 309:481-484.
- Kannan K, Jain SK (2000) Oxidative stress and apoptosis. Pathophysiology 7:153-163.
- Yang Y, Ma Z, Hu W, Wang D, Jiang S, Fan C, Di S, Liu D, Sun Y, Yi W (2016) Caveolin-1/-3: therapeutic targets for myocardial ischemia/reperfusion injury. Basic Res Cardiol 111:45.
- Motterlini R, Foresti R, Bassi R, Green CJ (2000) Curcumin, an antioxidant and anti-inflammatory agent, induces heme oxygenase-1 and protects endothelial cells against oxidative stress. Free Radic Biol Med 28:1303-1312.
- Barski OA, Tipparaju SM, Bhatnagar A (2008) The aldo-keto reductase superfamily and its role in drug metabolism and detoxification. Drug Metab Rev 40:553-624.
- Srivastava SK, Yadav UC, Reddy AB, Saxena A, Tammali R, Shoeb M, Ansari NH, Bhatnagar A, Petrash MJ, Srivastava S, Ramana KV (2011) Aldose reductase inhibition suppresses oxidative stress-induced inflammatory disorders. Chem Biol Interact 191:330-338.
- Lyon RC, Li D, McGarvie G, Ellis EM (2013) Aldo-keto reductases mediate constitutive and inducible protection against aldehyde toxicity in human neuroblastoma SH-SY5Y cells. Neurochem Int 62:113-121.
- Pladzyk A, Ramana KV, Ansari NH, Srivastava SK (2006) Aldose reductase prevents aldehyde toxicity in cultured human lens epithelial cells. Exp Eye Res 83:408-416.
- Wadia JS, Dowdy SF (2002) Protein transduction technology. Curr Opin Biotechnol 13:52-56.
- Dietz GP (2010) Cell-penetrating peptide technology to deliver chaperones and associated factors in diseases and basic research. Curr Pharm Biotechnol 11:167-174.
- van den Berg A, Dowdy SF (2011) Protein transduction domain delivery of therapeutic macromolecules. Curr Opin Biotechnol 22:888-893.
- Kim HR, Kim DW, Jo HS, Cho SB, Park JH, Lee CH, Choi YJ, Yeo EJ, Park SY, Kim ST, Yu YH, Kim DS, Kim HA, Cho SW, Han KH, Park J, Eum WS, Choi SY (2015) Tat-biliverdin reductase A inhibits inflammatory response by regulation of MAPK and NF-κB pathways in Raw 264.7 cells and edema mouse model. Mol Immunol 63:355-366.
- Shin MJ, Kim DW, Lee YP, Ahn EH, Jo HS, Kim DS, Kwon OS, Kang TC, Cho YJ, Park J, Eum WS, Choi SY (2014) Tat-glyoxalase protein inhibits against ischemic neuronal cell damage and ameliorates ischemic injury. Free Radic Biol Med 67:195-210.
- Shin MJ, Kim DW, Jo HS, Cho SB, Park JH, Lee CH, Yeo EJ, Choi YJ, Kim JA, Hwang JS, Sohn EJ, Jeong JH, Kim DS, Kwon HY, Cho YJ, Lee K, Han KH, Park J, Eum WS, Choi SY (2016) Tat-PRAS40 prevent hippocampal HT-22 cell death and oxidative stress induced animal brain ischemic insults. Free Radic Biol Med 97:250-262.
- Kim YN, Jung HY, Eum WS, Kim DW, Shin MJ, Ahn EH, Kim SJ, Lee CH, Yong JI, Ryu EJ, Park J, Choi JH, Hwang IK, Choi SY (2014) Neuroprotective effects of PEP-1-carbonyl reductase 1 against oxidative-stress-induced ischemic neuronal cell damage. Free Radic Biol Med 69:181-196.
- Kim MJ, Kim DW, Park JH, Kim SJ, Lee CH, Yong JI, Ryu EJ, Cho SB, Yeo HJ, Hyeon J, Cho SW, Kim DS, Son O, Park J, Han KH, Cho YS, Eum WS, Choi SY (2013) PEP-1-SIRT2 inhibits inflammatory response and oxidative stress-induced cell death via expression of antioxidant enzymes in murine macrophages. Free Radic Biol Med 63:432-445.
- Kim DW, Shin MJ, Choi YJ, Kwon HJ, Lee SH, Lee S, Park J, Han KH, Eum WS, Choi SY (2018) Tat-ATOX1 inhibits inflammatory responses via regulation of MAPK and NF-κB pathways. BMB Rep 51:654-659.
- Lee SJ, Kang HK, Choi YJ, Eum WS, Park J, Choi SY, Kwon HY (2018) PEP-1-paraoxonase 1 fusion protein prevents cytokine- induced cell destruction and impaired insulin secretion in rat insulinoma cells. BMB Rep 51:538-543.
- Yeo H, Yeo EJ, Shin MJ, Choi YJ, Lee CH, Kwon HY, Kim DW, Eum WS, Choi SY (2018) Protective effects of Tat-DJ-1 protein against streptozotocin-induced diabetes in a mice model. BMB Rep 51:362-367.
- Bradford MM (1976) A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem 72:248-254.
- Kang KT, Kwon YW, Kim DK, Lee SI, Kim KH, Suh DS, Kim JH (2018) TRRAP stimulates the tumorigenic potential of ovarian cancer stem cells. BMB Rep 51:514-519.
- Son Y, Kim S, Chung HT, Pae HO (2013) Reactive oxygen species in the activation of MAP kinases. Methods Enzymol 528:27-48.
- Hattangadi DK, DeMasters GA, Walker TD, Jones KR, Di X, Newsham IF, Gewirtz DA (2004) Influence of p53 and caspase 3 activity on cell death and senescence in response to methotrexate in the breast tumor cell. Biochem Pharmacol 68:1699-1708.
- Jin Z, El-Deiry WS (2005) Overview of cell death signaling pathways. Cancer Biol Ther 4:139-163.
- Yamaguchi H, Chen J, Bhalla K, Wang HG (2004) Regulation of Bax activation and apoptotic response to microtubuledamaging agents by p53 transcription-dependent and -independent pathways. J Biol Chem 279:39431-39437.
- Tan J, Zhuang L, Leong HS, Iyer NG, Liu ET, Yu Q (2005) Pharmacologic modulation of glycogen synthase kinase- 3beta promotes p53-dependent apoptosis through a direct Bax-mediated mitochondrial pathway in colorectal cancer cells. Cancer Res 65:9012-9020.
- Ryter SW, Kim HP, Hoetzel A, Park JW, Nakahira K, Wang X, Choi AM (2007) Mechanisms of cell death in oxidative stress. Antioxid Redox Signal 9:49-89.
- Madamanchi NR, Vendrov A, Runge MS (2005) Oxidative stress and vascular disease. Arterioscler Thromb Vasc Biol 25:29-38.
- Andersen JK (2004) Oxidative stress in neurodegeneration: cause or consequence? Nat Med 10 Suppl:S18-S25.
- Cooke MS, Evans MD, Dizdaroglu M, Lunec J (2003) Oxidative DNA damage: mechanisms, mutation, and disease. FASEB J 17:1195-1214.
- Ramana KV, Fadl AA, Tammali R, Reddy AB, Chopra AK, Srivastava SK (2006) Aldose reductase mediates the lipopolysaccharide- induced release of inflammatory mediators in RAW264.7 murine macrophages. J Biol Chem 281:33019-33029.
- Lim KS, Won YW, Park YS, Kim YH (2010) Preparation and functional analysis of recombinant protein transduction domain-metallothionein fusion proteins. Biochimie 92:964-970.
- Schwarze SR, Ho A, Vocero-Akbani A, Dowdy SF (1999) In vivo protein transduction: delivery of a biologically active protein into the mouse. Science 285:1569-1572.
- Rao KS, Reddy MK, Horning JL, Labhasetwar V (2008) TATconjugated nanoparticles for the CNS delivery of anti-HIV drugs. Biomaterials 29:4429-4438.
- Kang ES, Iwata K, Ikami K, Ham SA, Kim HJ, Chang KC, Lee JH, Kim JH, Park SB, Kim JH, Yabe-Nishimura C, Seo HG (2011) Aldose reductase in keratinocytes attenuates cellular apoptosis and senescence induced by UV radiation. Free Radic Biol Med 50:680-688.
- Yabe-Nishimura C, Nishinaka T, Iwata K, Seo HG (2003) Upregulation of aldose reductase by the substrate, methylglyoxal. Chem Biol Interact 143-144:317-323.
- Chen QM, Prowse KR, Tu VC, Purdom S, Linskens MH (2001) Uncoupling the senescent phenotype from telomere shortening in hydrogen peroxide-treated fibroblasts. Exp Cell Res 265:294-303.
- d'Adda di Fagagna F, Reaper PM, Clay-Farrace L, Fiegler H, Carr P, Von Zglinicki T, Saretzki G, Carter NP, Jackson SP (2003) A DNA damage checkpoint response in telomereinitiated senescence. Nature 426:194-198.
- Chen H, Yoshioka H, Kim GS, Jung JE, Okami N, Sakata H, Maier CM, Narasimhan P, Goeders CE, Chan PH (2011) Oxidative stress in ischemic brain damage: mechanisms of cell death and potential molecular targets for neuroprotection. Antioxid Redox Signal 14:1505-1517.
- Won MH, Kang TC, Jeon GS, Lee JC, Kim DY, Choi EM, Lee KH, Choi CD, Chung MH, Cho SS (1999) Immunohistochemical detection of oxidative DNA damage induced by ischemia-reperfusion insults in gerbil hippocampus in vivo. Brain Res 836:70-78.
- Floyd RA (1990) Role of oxygen free radicals in carcinogenesis and brain ischemia. FASEB J 4:2587-2597.
- Pradeep H, Diya JB, Shashikumar S, Rajanikant GK (2012) Oxidative stress--assassin behind the ischemic stroke. Folia Neuropathol 50:219-230.
- Chen CH, Budas GR, Churchill EN, Disatnik MH, Hurley TD, Mochly-Rosen D (2008) Activation of aldehyde dehydrogenase- 2 reduces ischemic damage to the heart. Science 321:1493-1495.
- Cherubini A, Ruggiero C, Polidori MC, Mecocci P (2005) Potential markers of oxidative stress in stroke. Free Radic Biol Med 39:841-852.
- Keith RJ, Haberzettl P, Vladykovskaya E, Hill BG, Kaiserova K, Srivastava S, Barski O, Bhatnagar A (2009) Aldose reductase decreases endoplasmic reticulum stress in ischemic hearts. Chem Biol Interact 178:242-249.
- Kim SM, Hwang IK, Yoo DY, Eum WS, Kim DW, Shin MJ, Ahn EH, Jo HS, Ryu EJ, Yong JI, Cho SW, Kwon OS, Lee KW, Cho YS, Han KH, Park J, Choi SY (2015) Tat-antioxidant 1 protects against stress-induced hippocampal HT-22 cells death and attenuate ischaemic insult in animal model. J Cell Mol Med 19:1333-1345.
- Srivastava S, Chandra A, Wang LF, Seifert WE Jr, DaGue BB, Ansari NH, Srivastava SK, Bhatnagar A (1998) Metabolism of the lipid peroxidation product, 4-hydroxy-trans-2-nonenal, in isolated perfused rat heart. J Biol Chem 273:10893-10900.
- Srivastava S, Chandra A, Ansari NH, Srivastava SK, Bhatnagar A (1998) Identification of cardiac oxidoreductase(s) involved in the metabolism of the lipid peroxidation-derived aldehyde-4-hydroxynonenal. Biochem J 329:469-475.
- Choudhary S, Xiao T, Srivastava S, Zhang W, Chan LL, Vergara LA, Van Kuijk FJ, Ansari NH (2005) Toxicity and detoxification of lipid-derived aldehydes in cultured retinal pigmented epithelial cells. Toxicol Appl Pharmacol 204:122-134.
- Lee WC, Wong HY, Chai YY, Shi CW, Amino N, Kikuchi S, Huang SH (2012) Lipid peroxidation dysregulation in ischemic stroke: plasma 4-HNE as a potential biomarker? Biochem Biophys Res Commun 425:842-847.
- Shinmura K, Bolli R, Liu SQ, Tang XL, Kodani E, Xuan YT, Srivastava S, Bhatnagar A (2002) Aldose reductase is an obligatory mediator of the late phase of ischemic preconditioning. Circ Res 91:240-246.
- Cho JH, Hwang IK, Yoo KY, Kim SY, Kim DW, Kwon YG, Choi SY, Won MH (2008) Effective delivery of Pep-1-cargo protein into ischemic neurons and long-term neuroprotection of Pep-1-SOD1 against ischemic injury in the gerbil hippocampus. Neurochem Int 52:659-668.
- Cai SR, Xu G, Becker-Hapak M, Ma M, Dowdy SF, McLeod HL (2006) The kinetics and tissue distribution of protein transduction in mice. Eur J Pharm Sci 27:311-319.
- Jo HS, Kim DW, Shin MJ, Cho SB, Park JH, Lee CH, Yeo EJ, Choi YJ, Yeo HJ, Sohn EJ, Son O, Cho SW, Kim DS, Yu YH, Lee KW, Park J, Eum WS, Choi SY (2017) Tat-HSP22 inhibits oxidative stress-induced hippocampal neuronal cell death by regulation of the mitochondrial pathway. Mol Brain 10:1.
- Jo HS, Kim DS, Ahn EH, Kim DW, Shin MJ, Cho SB, Park JH, Lee CH, Yeo EJ, Choi YJ, Yeo HJ, Chung CS, Cho SW, Han KH, Park J, Eum WS, Choi SY (2016) Protective effects of Tat- NQO1 against oxidative stress-induced HT-22 cell damage, and ischemic injury in animals. BMB Rep 49:617-622.
- Pekny M, Nilsson M (2005) Astrocyte activation and reactive gliosis. Glia 50:427-434.
- Ito D, Tanaka K, Suzuki S, Dembo T, Fukuuchi Y (2001) Enhanced expression of Iba1, ionized calcium-binding adapter molecule 1, after transient focal cerebral ischemia in rat brain. Stroke 32:1208-1215.
- Aguzzi A, Barres BA, Bennett ML (2013) Microglia: scapegoat, saboteur, or something else? Science 339:156-161.
- Chen Z, Trapp BD (2016) Microglia and neuroprotection. J Neurochem 136 Suppl 1:10-17.
- Condello C, Yuan P, Grutzendler J (2018) Microglia-mediated neuroprotection, TREM2 and Alzheimer’s disease: evidence from optical imaging. Biol Psychiatry 83:377-387.
- Lee Y, Lee SR, Choi SS, Yeo HG, Chang KT, Lee HJ (2014) Therapeutically targeting neuroinflammation and microglia after acute ischemic stroke. Biomed Res Int 2014:297241.
- Wood PL (1995) Microglia as a unique cellular target in the treatment of stroke: potential neurotoxic mediators produced by activated microglia. Neurol Res 17:242-248.
- Jin R, Yang G, Li G (2010) Inflammatory mechanisms in ischemic stroke: role of inflammatory cells. J Leukoc Biol 87:779-789.
- Neumann H, Kotter MR, Franklin RJ (2009) Debris clearance by microglia: an essential link between degeneration and regeneration. Brain 132:288-295.
- Spencer NG, Schilling T, Miralles F, Eder C (2016) Mechanisms underlying interferon-γ-induced priming of microglial reactive oxygen species production. PLoS One 11:e0162497.
- Kraft AD, Harry GJ (2011) Features of microglia and neuroinflammation relevant to environmental exposure and neurotoxicity. Int J Environ Res Public Health 8:2980-3018.
- D Dello Russo C, Lisi L, Tentori L, Navarra P, Graziani G, Combs CK (2017) Exploiting microglial functions for the treatment of glioblastoma. Curr Cancer Drug Targets 17:267-281.
- Zhu J, Jiang L, Liu Y, Qian W, Liu J, Zhou J, Gao R, Xiao H, Wang J (2015) MAPK and NF-κB pathways are involved in bisphenol A-induced TNF-α and IL-6 production in BV2 microglial cells. Inflammation 38:637-648.
- Clausen BH, Lambertsen KL, Babcock AA, Holm TH, Dagnaes- Hansen F, Finsen B (2008) Interleukin-1beta and tumor necrosis factor-alpha are expressed by different subsets of microglia and macrophages after ischemic stroke in mice. J Neuroinflammation 5:46.
- Yasuda Y, Shimoda T, Uno K, Tateishi N, Furuya S, Tsuchihashi Y, Kawai Y, Naruse S, Fujita S (2011) Temporal and sequential changes of glial cells and cytokine expression during neuronal degeneration after transient global ischemia in rats. J Neuroinflammation 8:70.
- Espinosa-Garcia C, Sayeed I, Yousuf S, Atif F, Sergeeva EG, Neigh GN, Stein DG (2017) Stress primes microglial polarization after global ischemia: Therapeutic potential of progesterone. Brain Behav Immun 66:177-192.
- Yan A, Cai G, Xia W, Fu Y (2017) Thromboxane A2 receptor antagonist SQ29548 suppresses the LPS-induced release of inflammatory cytokines in BV2 microglia cells via suppressing MAPK and NF-κB signaling pathways. Mol Med Rep 16:2491-2496.
- Dong P, Ji X, Han W, Han H (2019) Oxymatrine exhibits antineuroinflammatory effects on Aβ1-42-induced primary microglia cells by inhibiting NF-κB and MAPK signaling pathways. Int Immunopharmacol 74:105686.
- Chu K, Yin B, Wang J, Peng G, Liang H, Xu Z, Du Y, Fang M, Xia Q, Luo B (2012) Inhibition of P2X7 receptor ameliorates transient global cerebral ischemia/reperfusion injury via modulating inflammatory responses in the rat hippocampus. J Neuroinflammation 9:69.
- Kim H, Ahn JH, Song M, Kim DW, Lee TK, Lee JC, Kim YM, Kim JD, Cho JH, Hwang IK, Yan BC, Won MH, Park JH (2019) Pretreated fucoidan confers neuroprotection against transient global cerebral ischemic injury in the gerbil hippocampal CA1 area via reducing of glial cell activation and oxidative stress. Biomed Pharmacother 109:1718-1727.
- Hur J, Lee P, Kim MJ, Kim Y, Cho YW (2010) Ischemiaactivated microglia induces neuronal injury via activation of gp91phox NADPH oxidase. Biochem Biophys Res Commun 391:1526-1530.
- Ha SK, Moon E, Ju MS, Kim DH, Ryu JH, Oh MS, Kim SY (2012) 6-Shogaol, a ginger product, modulates neuroinflammation: a new approach to neuroprotection. Neuropharmacology 63:211-223.
- Hamby AM, Suh SW, Kauppinen TM, Swanson RA (2007) Use of a poly(ADP-ribose) polymerase inhibitor to suppress inflammation and neuronal death after cerebral ischemiareperfusion. Stroke 38(2 Suppl):632-636.
- Yrjänheikki J, Tikka T, Keinänen R, Goldsteins G, Chan PH, Koistinaho J (1999) A tetracycline derivative, minocycline, reduces inflammation and protects against focal cerebral ischemia with a wide therapeutic window. Proc Natl Acad Sci U S A 96:13496-13500.
- Sun HJ, Xue DD, Lu BZ, Li Y, Sheng LX, Zhu Z, Zhou JX, Lin GJ, Lin SZ, Yan GM, Chen YP, Yin W (2019) A novel synthetic steroid of 2β,3α,5α-trihydroxy-androst-6-one alleviates the loss of rat retinal ganglion cells caused by acute intraocular hypertension via inhibiting the inflammatory activation of microglia. Molecules 24:E252.
- Sapkota A, Gaire BP, Cho KS, Jeon SJ, Kwon OW, Jang DS, Kim SY, Ryu JH, Choi JW (2017) Eupatilin exerts neuroprotective effects in mice with transient focal cerebral ischemia by reducing microglial activation. PLoS One 12:e0171479.
- Widmann C, Gandin C, Petit-Paitel A, Lazdunski M, Heurteaux C (2018) The Traditional Chinese Medicine MLC901 inhibits inflammation processes after focal cerebral ischemia. Sci Rep 8:18062.
- Luo Y, Wang C, Li WH, Liu J, He HH, Long JH, Yang J, Sui X, Wang S, You Z, Wang YA (2018) Madecassoside protects BV2 microglial cells from oxygen-glucose deprivation/reperfusion- induced injury via inhibition of the toll-like receptor 4 signaling pathway. Brain Res 1679:144-154.