Articles

Article Tools
 View Full Text View Full Text |
 Abstract Abstract |
 Article as PDF Article as PDF |
 Print this Article Print this Article |
 Pubmed Pubmed |
 PMC PMC |
 PubReader PubReader |
 Export to Citation Export to Citation |
 Email Alerts Email Alerts |
 Open Access Open Access |
Share this article on :



Stats or Metrics
Article
Original Article
Exp Neurobiol 2019; 28(2): 216-228
Published online April 30, 2019
https://doi.org/10.5607/en.2019.28.2.216
© The Korean Society for Brain and Neural Sciences
C3a Receptor Inhibition Protects Brain Endothelial Cells Against Oxygen-glucose Deprivation/Reperfusion
Saif Ahmad1*, Adam Kindelin1, Shah Alam Khan1,2, Maaz Ahmed1, Md Nasrul Hoda3, Kanchan Bhatia1,4, and Andrew F. Ducruet1*
1Department of Neurosurgery, Barrow Neurological Institute, St. Joseph's Hospital and Medical Center, Dignity Health, Phoenix, Arizona 85013, USA.
2Oman Medical College, Muscat 130, Sultanate of Oman.
3Department of Neurology, Barrow Neurological Institute, St. Joseph's Hospital and Medical Center, Dignity Health, Phoenix, Arizona 85013, USA.
4School of Mathematical and Natural Sciences, Arizona State University, Phoenix, AZ 85004, USA.
Correspondence to: *To whom correspondence should be addressed.
TEL: 1-602-406-3220, FAX: 1-602-406-4172
Saif Ahmad, e-mail: saif.ahmad@barrowneuro.org
Andrew F. Ducruet, e-mail: andrew.ducruet@barrowbrainandspine.com
Abstract
The complement cascade is a central component of innate immunity which plays a critical role in brain inflammation. Complement C3a receptor (C3aR) is a key mediator of post-ischemic cerebral injury, and pharmacological antagonism of the C3a receptor is neuroprotective in stroke. Cerebral ischemia injures brain endothelial cells, causing blood brain barrier (BBB) disruption which further exacerbates ischemic neuronal injury. In this study, we used an
Graphical Abstract
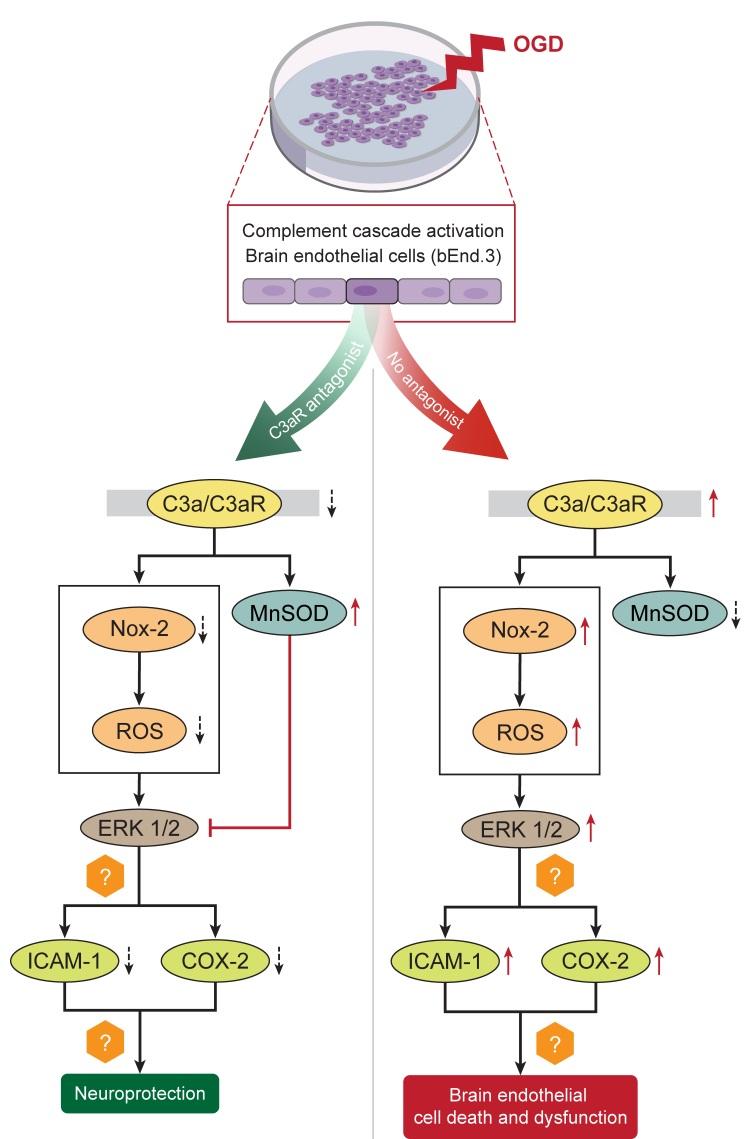
Keywords: bEnd.3 cells, Ischemia, OGD, Inflammation, Oxidative stress
INTRODUCTION
Ischemic stroke is the third leading cause of death and the leading cause of disability worldwide and claims around 130,000 lives annually. Ischemic stroke causes loss of brain function due to a reduction in cerebral blood flow which suppresses oxygen and glucose levels in the brain. This in turn triggers neuronal cell death and blood brain barrier (BBB) dysfunction via activation of inflammatory and cytotoxic molecules and oxidative stress signaling [1,2]. Ischemia is a major cause of brain endothelial cell injury and underlies several vascular diseases including atherosclerosis, hemorrhage, brain edema, and vascular dementia [3,4,5,6]. Furthermore, post-ischemic reperfusion, whether mechanical or pharmacologic, promotes secondary injury in part through increased inflammation and generation of free radicals [3,7,8,9]. Brain endothelial cells are one of the primary targets of this secondary injury that promotes vascular dysfunction, including blood brain barrier (BBB) dysfunction [10,11] and disruption of tight junctions that limit the passage of drugs from blood to brain [2]. Effective strategies to protect the vasculature against brain ischemia are still lacking.
The complement cascade, a component of innate immunity, is a critical effector of the immune response [12]. Complement activation has been implicated in several autoimmune and rheumatologic diseases, as well as stroke [13,14,15]. In stroke, complement activation promotes both local and systemic inflammation, triggering a wide range of processes that contribute to stroke severity and outcome [16,17,18]. Neurons and glia have been shown to produce complement proteins including C1q, C3, C3a, C5, and C5a [12,19,20]. Many reports have suggested that inhibition of complement activation is beneficial by suppressing post-ischemic inflammation and reducing infarct volume in animal models of stroke [15,21,22,23]. The complement anaphylatoxins C3a and C5a, potent inflammatory mediators which function through their cognate receptors, C3a (C3aR) and C5a (C5aR), have been implicated in leukocyte activation, vascular permeability, and chemotaxis [24,25]. Additionally, the C3a receptor plays a critical role in the pathophysiology of ischemia/reperfusion injury in diverse experimental models [21,22,26,27]. Multiple studies have shown that cerebral C3aR expression was increased in mouse and rat model of stroke, and C3aR antagonism improved outcome [22,28,29]. Further, it was reported that the complement C3a and its receptor is involved in brain endothelial activation [30], and the brain microvascular endothelial cells stimulated by tumor necrosis factor (TNF) exhibit C3aR activation [31].
In the present study, we utilized a model of murine brain microvascular endothelial cells (b.End.3) subjected to oxygen-glucose deprivation (OGD) followed by reperfusion to investigate the expression of C3aR as well as the beneficial effect of C3aR antagonist (C3aRA) administration on cell death, inflammation, oxidative stress, and expression of tight junction proteins.
MATERIALS AND METHODS
C3aR inhibitor (SB290157) was purchased from (Calbiochem, Darmstadt, Germany). Immortalized bEnd.3 cells were purchased from American Type Culture Collection (ATCC, Manassas, VA, USA). Dulbecco's Modified Eagle Medium (DMEM/High Glucose, GE, Utah, USA), Pen strep (Gibco, USA), FBS (VWR, USA) and all reagents used were AR and molecular grade. TUNEL assay kit was obtained from AAT Bioquest (USA). Antibodies (monoclonal anti-mouse Caspase3; monoclonal anti-mouse C3aR; monoclonal anti-mouse ICAM-1; monoclonal anti-mouse occludin; monoclonal anti-mouse Cox-2; monoclonal gp91-phox and MnSOD) were obtained from Santa Cruz Biotechnology, CA, USA. Recombinant C3a (rmC3a) was obtained from R&D system (USA). Lectin was purchased from Vector Lab (USA). Protease inhibitor cocktail was purchased from Sigma (MO). RIPA extraction buffer was procured from Fisher Scientific (USA). All secondary fluorescent antibodies were purchased from Invitrogen (USA).
Endothelial cell culture was performed as previously described [32]. In brief, the immortalized mouse brain endothelial cell line (bEnd.3) was cultured in DMEM with high glucose (4.5 mg/ml) supplemented with 10% FBS, and antibiotic. The cells were incubated in a CO2 incubator with 5% CO2 at 37℃. OGD is used as an
Apoptotic cell death was analyzed using a terminal deoxynucleotidyl transferase-mediated uridine 5′-triphosphate-biotin nick end labeling (TUNEL) assay kit (In Situ Cell death Detection Fluorescein, Roche, Germany). In brief, bEnd.3 cells were grown on glass coverslips and after C3aRA treatment in the setting of OGD/reperfusion, cells were fixed with 4% PFA and then washed with 1X PBS three times. The fixed cells were permeabilized with 0.1% triton X-100 (PBS-T) for 5 minutes before incubating with fluorescein dUTP TUNEL reaction mixture for 1 h at 37℃. After the PBS wash, cells were counterstained with DAPI (Vector Laboratories, USA). The slides were evaluated under the fluorescence microscope (Leica, Germany) and TUNEL positive nuclei were counted by researcher blinded to treatment. The experiment was repeated 3 times in the same environment and images were taken from 4 non-overlapping fields of coverslips. The percentage of TUNEL positive cells relative to the total number of cell nuclei counterstained with DAPI was used to calculate the TUNEL positive ratio (number of green nuclei/number of blue nuclei) × 100%.
bEnd.3 cells were plated on coverslips in 6 well plates (Corning, USA) and then subjected to OGD as described above. Cells were fixed using 4% PFA for 20 minutes at room temperature. For immunofluorescence study, samples were blocked with 10% goat serum in TBS with 0.5% Triton X-100 for 1 hr. after the washing with PBS-T (0.1% Triton X-100), primary antibodies (Caspase-3, C3aR, ICAM-1 and Cox-2; Santa Cruz Biotechnology, 1:50 dilution) were added and incubated overnight at 4℃. Cells were washed 3 times with PBS-T and then again incubated in fluorescein anti-mouse secondary antibodies (Invitrogen, 1:200), mounted with DAPI (Vector), and analyzed using fluorescence microscope (Keyence) by using 20 and 40 × objectives. Fluorescence intensity was measured by Image J software (NIH) [33].
The bEnd.3 cells were homogenized in modified RIPA buffer (Upstate, Lake Placid, NY), containing 50 mM Tris, 150 mM NaCl, 1 mM EDTA, 1% Nonidet P-40, 0.25% deoxycholate, supplemented with 40 mM NaF, 2 mM Na3VO4, 0.5 mM phenylmethylsulfonyl fluoride and 1:100 (v/v) of proteinase inhibitor cocktail (Sigma). Insoluble material was removed by centrifugation at 12,000 ×g at 4℃ for 30 min. Protein concentration was determined by DC Protein Assay (Bio-Rad, Hercules, CA). Equal amount of protein was loaded on SDS-PAGE (gradient gel - 4 to 20%; Pierce, Rockford, IL) and subsequently transferred to PVDF membrane (Biorad) and probed with specific antibodies (anti-mouse C3aR, Occludin, gp91-phox and MnSOD; 1:1000) overnight followed by 3 washes with TBS-T. Membranes were then incubated with secondary antibodies HRP conjugated for 1 h at RT and, after 3 washes with TBS, bands were visualized using the enhanced Amersham ECL western blotting detection kit (GE Healthcare, USA). Protein expression was detected as previously described [34]. The same membranes were re-probed using β-actin as an internal control. Quantification of immunoreactivity was performed using densitometry analysis in the standard fashion (Image J software/NIH) [33].
Quantitative data are presented as mean±SEM. Differences among experimental groups were analyzed by analysis of variance (one-way ANOVA) followed by Newman-Keuls multiple comparison tests. Significance was defined as p<0.05. Data were analyzed using Graph Pad PRISM software.
RESULTS
In the present study, OGD led to a significant increase in C3aR protein expression level (22.2%, vs. 10.6% control) in bEnd.3 cells relative to control. Interestingly, C3aR antagonist administration (C3aRA; SB290157) significantly reduced (14.2%) the expression of C3aR protein compared following OGD (Fig. 1A~D).
OGD promotes apoptotic cell death in bEND3 cells [2]. To determine whether C3aR inhibition attenuates cell death, we investigated the effect of OGD on endothelial apoptosis using TUNEL assay and Caspase-3 staining (Fig. 2A~D). Both TUNEL and Caspase-3 staining showed increased apoptotic cell death (23.8% vs. 7.2% control) against OGD when compared with control group. C3aRA treatment significantly reduced the cell death (16.0%) subjected to OGD.
ICAM-1 induces intracellular signaling in endothelial cells and its increased expression causes actin cytoskeleton reorganization, endothelial cell leakiness and junctional alterations [35]. OGD promotes ICAM-1 upregulation in bEnd.3 cells as compared with control. We found that C3aRA treatment significantly attenuates endothelial ICAM-1 expression. Double staining of ICAM-1 and C3aR demonstrates the co-localization of both the proteins in endothelial cells. Next, we observed a significant upregulation of endothelial Cox-2 expression following OGD which suppressed by C3aRA treatment. Upregulation of Cox-2, a rate limiting key enzyme in the synthesis of prostaglandins, mediates brain vascular inflammation [36]. These results suggest that C3aRA significantly attenuates expression of endothelial inflammatory markers following OGD which reflects decreased inflammation (Fig. 3A~C & Fig. 4A and 4B).
NADPH oxidase subunit gp91phox (Nox2) contributes to endothelial dysfunction and permeability and is activated by hypoxia [37]. On the other hand, the antioxidant enzyme MnSOD plays a prominent protective role in vascular inflammation [38]. Here we demonstrated that OGD significantly elevated gp91-phox expression (19.9% vs. 11% control) and decreased MnSOD level (10.8% vs. 23.1% control) in bEnd.3 cells compared with controls. C3aRA treatment attenuated the expression of gp91-phox (15.8% vs. OGD) and MnSOD (19.8% vs. OGD) following OGD respectively (Fig. 5A~D). These data suggest that C3aR antagonism reduces oxidative stress and may improve antioxidant activity following experimental OGD.
Tight junction proteins including ZO-1 and occludin play a key role in the maintenance of BBB integrity [39]. We observed decreased occludin expression (68% vs. Control) in endothelial cells subjected to OGD relative to controls. Administration of C3aRA restored occludin expression (54% vs. OGD) on following OGD (Fig. 6A and 6B). ERK1/2 activation has been reported to promote cell death in ischemia/reperfusion injury models [9]. Prior evidence has shown that C3aR activation leads to signaling mediated through pERK [40]. We therefore sought to evaluate levels of pERK in our model to demonstrate that endothelial C3aR inhibition functions through mediation of downstream pERK. We observed an increased expression of pERK1/2 (68% vs. control) in endothelial cells subjected to OGD relative to controls. Administration of C3aRA attenuated the expression of pERK1/2 (42% vs. OGD) following OGD (Fig. 6C and 6D).
To determine the effect of exogenous C3a on bENd3 cells following OGD, we found that C3a administration alone increased the expression of Caspase3, ICAM-1 and pERK. We also found that C3aRA in the presence of C3a significantly reduced the expression level of these markers following OGD (Fig. 7A~H).
DISCUSSION
In the present study, we show that C3aR inhibition reduces endothelial cell death, attenuates inflammatory and oxidative stress signaling, and restores the expression of tight junction proteins following OGD. Cerebral microvascular endothelial cell (bEnd.3) culture represents an excellent model to study brain endothelial cell vascular dysfunction, and OGD/hypoxia activates inflammatory and oxidative stress mechanisms and degrades endothelial cell barrier function [10,16,41,42,43].
The complement system is a fundamental component of innate immunity and complement activation mediates ischemia/reperfusion injury across diverse organ systems [22,44]. Endothelial expression of complement C3a receptor induces a variety of cellular responses, including activation of endothelial cells and increased endothelial permeability [22,28]. Here, we investigated the protective role of C3aRA in an OGD induced endothelial cell culture model of experimental ischemia. In the present study, OGD promoted endothelial cell expression of the C3aR. Administration of the C3aRA reduced C3aR expression levels. This is likely the result of an indirect feedback effect due to reduced inflammatory signaling and endothelial cell activation through abrogation of C3aR signaling, and further work is necessary to characterize this mechanism. We also found that treatment with C3aRA attenuated apoptotic cell death and C3aR expression level in bEnd.3 cells subjected to OGD followed by reperfusion. Further, OGD significantly increased the expression of the inflammatory markers ICAM-1 and Cox-2, and this was expression was attenuated by C3aRA. Increased ICAM-1 expression has been shown to promote endothelial permeability, cytoskeletal reorganization and junctional alterations [35]. Cerebral endothelial ICAM-1 expression also promotes neutrophil infiltration through ischemic endothelium [22]. In the present study, C3aRA administration suppressed ICAM-1 expression, suggesting that that C3aR plays an upstream role in ICAM-1 upregulation in ischemic endothelial cells. This has been observed previously in a murine model of middle cerebral artery occlusion, where the C3aRA-treated group exhibited significant reductions in ICAM-1 expression [22]. In the present study, we also demonstrated cyclooxygenase-2 (Cox-2) upregulation in endothelial cells following OGD. Cox-2, a key enzyme in the biosynthesis of prostaglandins, is upregulated in endothelial cells following administration of LPS and VEGF [45,46]. Cox-2 serves a crucial role in the development of vascular inflammation [36]. Our findings suggest that complement C3aR may regulate post-ischemic endothelial Cox-2 expression and C3aR antagonism may be attenuating increased Cox-2 expression.
We further investigated the role of C3aRA on oxidative stress signaling pathways and observed that OGD increased NADPH oxidase subunit gp91-phox (Nox-2) expression. Several studies have suggested that ischemia/reperfusion generates increased reactive oxygen species (ROS) that in turn play a major role in endothelial cell (EC) activation and barrier function; and that the major source of ROS in endothelial cells is NADPH oxidase [47]. It is known that NADPH oxidase is activated by several factors including hypoxia, growth factors, cytokines and shear stress [48], and that Nox-2 is a critical component of endothelial NADPH oxidase [47]. Here, we found that C3aRA treatment significantly reduced Nox-2 expression in endothelial cells subjected to OGD, and our data is an agreement with a recent study showing that complement receptor 3 mediates NADPH oxidase activation [49]. On the other hand, we noted that mitochondrial superoxide dismutase (MnSOD) expression was suppressed in endothelial cells subjected to OGD and treatment with C3aRA restored its expression level significantly. There is a balance between intracellular ROS and antioxidant enzymes including SODs, whose main function is to convert O2 - to H2O2 [47]. MnSOD may participate in cellular defense in cerebral endothelial cells and there is profound MnSOD loss in cerebral endothelial cells due to oxidative stress [50]. Some reports suggest that MnSOD contributes in antioxidant defense in endothelial cells and the regulation of blood-brain barrier [51,52]. Our data demonstrate that C3aRA treatment protects cerebral endothelial cells by supporting the intracellular antioxidant defense system against OGD induced oxidative stress.
The blood-brain barrier (BBB) serves to maintain the homeostasis of the central nervous system (CNS), and endothelial cells along with glia and pericytes comprise this important structure. Endothelial cells which constitute the BBB exhibit a specific phenotype characterized by the presence of tight junctions (TJs) and the expression of a uniquely polarized transport system [53]. Brain endothelial TJs are composed of integral membrane proteins, including occludin, which are crucial to BBB function [53,54]. In the present study, we found that endothelial expression of occludin was suppressed following OGD and administration of C3aRA restored its expression. Similar results have been observed for expression of tight junction proteins in brain endothelial cells, which are downregulated by beta amyloid toxicity [43]. In another study, disruption of occludin expression in brain endothelial cells was reported following hypoxia/aglycemia [42]. Our results are in agreement with recent study in which hypoxia/OGD induces endothelial barrier dysfunction by downregulating tight junction proteins [41].
We further tested the hypothesis that C3aR activation functions through ERK1/2 pathway signaling in endothelial cells subjected to OGD. We found that the OGD significantly increased the phosphorylation of ERK1/2 and inhibition of C3aR attenuated the ERK1/2 expression level. It has been noted that ischemia induces activation of ERK1/2 signaling in cerebral endothelial cells which triggers endothelial cell death, and on the other hand ERK1 and ERK2 regulate endothelial cell proliferation and migration in angiogenesis [8,9]. Interestingly, NADPH oxidase has been reported to be involved in activation of ERK signaling, and ERK1/2 cascade activates Cox-2 and ICAM-1signaling in different disease models [49,55,56]. Moreover, SODs prevent ERK activation and cell death [9]. Our finding is an agreement with other studies which reported complement activation through C3a and C5a induces ERK1/2 signaling during ischemia/ reperfusion injury [57,58]. Together these data provide mechanistic insight into the fact that C3aR inhibition during ischemia/reperfusion may attenuate expression of Nox2/ICAM-1/Cox-2 and prevent endothelial cell death through ERK signaling. Next we administered exogenous C3a alone on bEnd3 cells with and without C3aRA following OGD. We found that C3a administration significantly increased endothelial cell expression level of Caspase-3, ICAM-1 and pERK in OGD compared with OGD alone. C3aRA administration reduced the expression level of these same markers both with and without C3a. C3a is a pro-inflammatory molecule and locally produced C3a interacts with its G-protein-coupled receptor C3aR and is involved in endothelial activation and increasing vascular permeability [30,31,59]. Our finding suggests that C3a contributes in cell death and inflammation in OGD and antagonism of the C3a receptor reduced its proinflammatory effect in brain endothelial cells.
In conclusion, the present study demonstrates a potential protective role for C3a receptor antagonist administration in cerebral endothelial cells subjected to experimental OGD through the attenuation of caspase-mediated apoptosis, inflammation, oxidative stress and ERK signaling (Fig. 8). C3aRA treatment also restores the expression of tight junction proteins and may have a beneficial effect on BBB function following experimental ischemia. These results contribute to our understanding of the function of C3aR in endothelial cells as well as the mechanisms underlying C3aRmediated neuroprotection in brain endothelial cells.
Figures

{kind=link}

{kind=link}

{kind=link}

{kind=link}
{kind=link}
{kind=link}

{kind=link}
{kind=link}
References
- Tornabene E, Brodin B. Stroke and drug delivery--
in vitro models of the ischemic blood-brain barrier. J Pharm Sci 2016;105:398-405.
- Zhang Y, Wang T, Yang K, Xu J, Ren L, Li W, Liu W. Cerebral microvascular endothelial cell apoptosis after ischemia: role of enolase-phosphatase 1 activation and aci-reductone dioxygenase 1 translocation. Front Mol Neurosci 2016;9:79.
- He Y, Luan Z, Fu X, Xu X. Overexpression of uncoupling protein 2 inhibits the high glucose-induced apoptosis of human umbilical vein endothelial cells. Int J Mol Med 2016;37:631-638.
- Tsai HY, Lin CP, Huang PH, Li SY, Chen JS, Lin FY, Chen JW, Lin SJ. Coenzyme Q10 attenuates high glucose-induced endothelial progenitor cell dysfunction through AMP-activated protein kinase pathways. J Diabetes Res 2016;2016:6384759.
- Li W, Chen Z, Yan M, He P, Chen Z, Dai H. The protective role of isorhamnetin on human brain microvascular endothelial cells from cytotoxicity induced by methylglyoxal and oxygen-glucose deprivation. J Neurochem 2016;136:651-659.
- Song J, Kang SM, Lee WT, Park KA, Lee KM, Lee JE. Glutathione protects brain endothelial cells from hydrogen peroxide-induced oxidative stress by increasing nrf2 expression. Exp Neurobiol 2014;23:93-103.
- Yang J, Ahn HN, Chang M, Narasimhan P, Chan PH, Song YS. Complement component 3 inhibition by an antioxidant is neuroprotective after cerebral ischemia and reperfusion in mice. J Neurochem 2013;124:523-535.
- Srinivasan R, Zabuawala T, Huang H, Zhang J, Gulati P, Fernandez S, Karlo JC, Landreth GE, Leone G, Ostrowski MC. Erk1 and Erk2 regulate endothelial cell proliferation and migration during mouse embryonic angiogenesis. PLoS One 2009;4:e8283.
- Narasimhan P, Liu J, Song YS, Massengale JL, Chan PH. VEGF Stimulates the ERK 1/2 signaling pathway and apoptosis in cerebral endothelial cells after ischemic conditions. Stroke 2009;40:1467-1473.
- Liao LX, Zhao MB, Dong X, Jiang Y, Zeng KW, Tu PF. TDB protects vascular endothelial cells against oxygenglucose deprivation/reperfusion-induced injury by targeting miR-34a to increase Bcl-2 expression. Sci Rep 2016;6:37959.
- Cecchelli R, Berezowski V, Lundquist S, Culot M, Renftel M, Dehouck MP, Fenart L. Modelling of the blood-brain barrier in drug discovery and development. Nat Rev Drug Discov 2007;6:650-661.
- Rutkowski MJ, Sughrue ME, Kane AJ, Mills SA, Fang S, Parsa AT. Complement and the central nervous system: emerging roles in development, protection and regeneration. Immunol Cell Biol 2010;88:781-786.
- Walport MJ. Complement. First of two parts. N Engl J Med 2001;344:1058-1066.
- Walport MJ. Complement. Second of two parts. N Engl J Med 2001;344:1140-1144.
- Ducruet AF, Zacharia BE, Sosunov SA, Gigante PR, Yeh ML, Gorski JW, Otten ML, Hwang RY, DeRosa PA, Hickman ZL, Sergot P, Connolly ES. Complement inhibition promotes endogenous neurogenesis and sustained antiinflammatory neuroprotection following reperfused stroke. PLoS One 2012;7:e38664.
- Khoyetsyan A, Kacimi R, Tsakanova G, Boyajyan A, Arakelyan A, Yenari MA. Activated complement protein C5a does not affect brain-derived endothelial cell viability and zonula occludens-1 levels following oxygen-glucose deprivation. Brain Circ 2017;3:14-20.
- Kvietys PR, Granger DN. Role of reactive oxygen and nitrogen species in the vascular responses to inflammation. Free Radic Biol Med 2012;52:556-592.
- Denes A, Thornton P, Rothwell NJ, Allan SM. Inflammation and brain injury: acute cerebral ischaemia, peripheral and central inflammation. Brain Behav Immun 2010;24:708-723.
- Morgan BP, Gasque P. Extrahepatic complement biosynthesis: where, when and why?. Clin Exp Immunol 1997;107:1-7.
- D'Ambrosio AL, Pinsky DJ, Connolly ES. The role of the complement cascade in ischemia/reperfusion injury: implications for neuroprotection. Mol Med 2001;7:367-382.
- Rynkowski MA, Kim GH, Garrett MC, Zacharia BE, Otten ML, Sosunov SA, Komotar RJ, Hassid BG, Ducruet AF, Lambris JD, Connolly ES. C3a receptor antagonist attenuates brain injury after intracerebral hemorrhage. J Cereb Blood Flow Metab 2009;29:98-107.
- Ducruet AF, Hassid BG, Mack WJ, Sosunov SA, Otten ML, Fusco DJ, Hickman ZL, Kim GH, Komotar RJ, Mocco J, Connolly ES. C3a receptor modulation of granulocyte infiltration after murine focal cerebral ischemia is reperfusion dependent. J Cereb Blood Flow Metab 2008;28:1048-1058.
- Atkinson C, Zhu H, Qiao F, Varela JC, Yu J, Song H, Kindy MS, Tomlinson S. Complement-dependent P-selectin expression and injury following ischemic stroke. J Immunol 2006;177:7266-7274.
- Drouin SM, Kildsgaard J, Haviland J, Zabner J, Jia HP, McCray PB, Tack BF, Wetsel RA. Expression of the complement anaphylatoxin C3a and C5a receptors on bronchial epithelial and smooth muscle cells in models of sepsis and asthma. J Immunol 2001;166:2025-2032.
- Gasque P, Singhrao SK, Neal JW, Wang P, Sayah S, Fontaine M, Morgan BP. The receptor for complement anaphylatoxin C3a is expressed by myeloid cells and nonmyeloid cells in inflamed human central nervous system: analysis in multiple sclerosis and bacterial meningitis. J Immunol 1998;160:3543-3554.
- Proctor LM, Arumugam TV, Shiels I, Reid RC, Fairlie DP, Taylor SM. Comparative anti-inflammatory activities of antagonists to C3a and C5a receptors in a rat model of intestinal ischaemia/reperfusion injury. Br J Pharmacol 2004;142:756-764.
- Wu MC, Brennan FH, Lynch JP, Mantovani S, Phipps S, Wetsel RA, Ruitenberg MJ, Taylor SM, Woodruff TM. The receptor for complement component C3a mediates protection from intestinal ischemia-reperfusion injuries by inhibiting neutrophil mobilization. Proc Natl Acad Sci U S A 2013;110:9439-9444.
- Barnum SR, Ames RS, Maycox PR, Hadingham SJ, Meakin J, Harrison D, Parsons AA. Expression of the complement C3a and C5a receptors after permanent focal ischemia: an alternative interpretation. Glia 2002;38:169-173.
- Van Beek J, Bernaudin M, Petit E, Gasque P, Nouvelot A, MacKenzie ET, Fontaine M. Expression of receptors for complement anaphylatoxins C3a and C5a following permanent focal cerebral ischemia in the mouse. Exp Neurol 2000;161:373-382.
- Wu F, Zou Q, Ding X, Shi D, Zhu X, Hu W, Liu L, Zhou H. Complement component C3a plays a critical role in endothelial activation and leukocyte recruitment into the brain. J Neuroinflammation 2016;13:23.
- Sartain SE, Turner NA, Moake JL. Brain microvascular endothelial cells exhibit lower activation of the alternative complement pathway than glomerular microvascular endothelial cells. J Biol Chem 2018;293:7195-7208.
- Zhao XJ, Larkin TM, Lauver MA, Ahmad S, Ducruet AF. Tissue plasminogen activator mediates deleterious complement cascade activation in stroke. PLoS One 2017;12:e0180822.
- Ahmad S, Elsherbiny NM, Haque R, Khan MB, Ishrat T, Shah ZA, Khan MM, Ali M, Jamal A, Katare DP, Liou GI, Bhatia K. Sesamin attenuates neurotoxicity in mouse model of ischemic brain stroke. Neurotoxicology 2014;45:100-110.
- Ahmad S, Wang Y, Shaik GM, Burghes AH, Gangwani L. The zinc finger protein ZPR1 is a potential modifier of spinal muscular atrophy. Hum Mol Genet 2012;21:2745-2758.
- Clark PR, Manes TD, Pober JS, Kluger MS. Increased ICAM-1 expression causes endothelial cell leakiness, cytoskeletal reorganization and junctional alterations. J Invest Dermatol 2007;127:762-774.
- Lin CC, Hsieh HL, Shih RH, Chi PL, Cheng SE, Yang CM. Up-regulation of COX-2/PGE2 by endothelin-1 via MAPK-dependent NF-κB pathway in mouse brain microvascular endothelial cells. Cell Commun Signal 2013;11:8.
- Frey RS, Ushio-Fukai M, Malik AB. NADPH oxidasedependent signaling in endothelial cells: role in physiology and pathophysiology. Antioxid Redox Signal 2009;11:791-810.
- Ungvari Z, Labinskyy N, Mukhopadhyay P, Pinto JT, Bagi Z, Ballabh P, Zhang C, Pacher P, Csiszar A. Resveratrol attenuates mitochondrial oxidative stress in coronary arterial endothelial cells. Am J Physiol Heart Circ Physiol 2009;297:H1876-H1881.
- Strazielle N, Ghersi-Egea JF. Physiology of blood-brain interfaces in relation to brain disposition of small compounds and macromolecules. Mol Pharm 2013;10:1473-1491.
- Li K, Fazekasova H, Wang N, Peng Q, Sacks SH, Lombardi G, Zhou W. Functional modulation of human monocytes derived DCs by anaphylatoxins C3a and C5a. Immunobiology 2012;217:65-73.
- Ma X, Zhang H, Pan Q, Zhao Y, Chen J, Zhao B, Chen Y. Hypoxia/aglycemia-induced endothelial barrier dysfunction and tight junction protein downregulation can be ameliorated by citicoline. PLoS One 2013;8:e82604.
- Brown RC, Davis TP. Hypoxia/aglycemia alters expression of occludin and actin in brain endothelial cells. Biochem Biophys Res Commun 2005;327:1114-1123.
- Song J, Choi SM, Whitcomb DJ, Kim BC. Adiponectin controls the apoptosis and the expression of tight junction proteins in brain endothelial cells through AdipoR1 under beta amyloid toxicity. Cell Death Dis 2017;8:e3102.
- Cowell RM, Plane JM, Silverstein FS. Complement activation contributes to hypoxic-ischemic brain injury in neonatal rats. J Neurosci 2003;23:9459-9468.
- Akarasereenont PC, Techatraisak K, Thaworn A, Chotewuttakorn S. The expression of COX-2 in VEGF-treated endothelial cells is mediated through protein tyrosine kinase. Mediators Inflamm 2002;11:17-22.
- Matsumura K, Cao C, Ozaki M, Morii H, Nakadate K, Watanabe Y. Brain endothelial cells express cyclooxygenase-2 during lipopolysaccharide-induced fever: light and electron microscopic immunocytochemical studies. J Neurosci 1998;18:6279-6289.
- Pendyala S, Usatyuk PV, Gorshkova IA, Garcia JG, Natarajan V. Regulation of NADPH oxidase in vascular endothelium: the role of phospholipases, protein kinases, and cytoskeletal proteins. Antioxid Redox Signal 2009;11:841-860.
- Griendling KK, Sorescu D, Ushio-Fukai M. NAD(P)H oxidase: role in cardiovascular biology and disease. Circ Res 2000;86:494-501.
- Hou L, Wang K, Zhang C, Sun F, Che Y, Zhao X, Zhang D, Li H, Wang Q. Complement receptor 3 mediates NADPH oxidase activation and dopaminergic neurodegeneration through a Src-Erk-dependent pathway. Redox Biol 2018;14:250-260.
- Méthy D, Bertrand N, Prigent-Tessier A, Stanimirovic D, Beley A, Marie C. Differential MnSOD and HO-1 expression in cerebral endothelial cells in response to sublethal oxidative stress. Brain Res 2004;1003:151-158.
- Liu C, Ogando D, Bonanno JA. SOD2 contributes to anti-oxidative capacity in rabbit corneal endothelial cells. Mol Vis 2011;17:2473-2481.
- Freeman LR, Keller JN. Oxidative stress and cerebral endothelial cells: regulation of the blood-brain-barrier and antioxidant based interventions. Biochim Biophys Acta 2012;1822:822-829.
- Luissint AC, Artus C, Glacial F, Ganeshamoorthy K, Couraud PO. Tight junctions at the blood brain barrier: physiological architecture and disease-associated dysregulation. Fluids Barriers CNS 2012;9:23.
- Pan R, Yu K, Weatherwax T, Zheng H, Liu W, Liu KJ. Blood occludin level as a potential biomarker for early blood brain barrier damage following ischemic stroke. Sci Rep 2017;7:40331.
- Lee SJ, Drabik K, Van Wagoner NJ, Lee S, Choi C, Dong Y, Benveniste EN. ICAM-1-induced expression of proinflammatory cytokines in astrocytes: involvement of extracellular signal-regulated kinase and p38 mitogen-activated protein kinase pathways. J Immunol 2000;165:4658-4666.
- Huang F, Cao J, Liu Q, Zou Y, Li H, Yin T. MAPK/ERK signal pathway involved expression of COX-2 and VEGF by IL-1β induced in human endometriosis stromal cells
in vitro . Int J Clin Exp Pathol 2013;6:2129-2136.
- Castellano G, Franzin R, Stasi A, Divella C, Sallustio F, Pontrelli P, Lucarelli G, Battaglia M, Staffieri F, Crovace A, Stallone G, Seelen M, Daha MR, Grandaliano G, Gesualdo L. Complement activation during ischemia/reperfusion injury induces pericyte-to-myofibroblast transdifferentiation regulating peritubular capillary lumen reduction through pERK signaling. Front Immunol 2018;9:1002.
- Kraus S, Seger R, Fishelson Z. Involvement of the ERK mitogen-activated protein kinase in cell resistance to complement-mediated lysis. Clin Exp Immunol 2001;123:366-374.
- Coulthard LG, Woodruff TM. Is the complement activation product C3a a proinflammatory molecule? Reevaluating the evidence and the myth. J Immunol 2015;194:3542-3548.