Articles

Article Tools
 View Full Text View Full Text |
 Abstract Abstract |
 Article as PDF Article as PDF |
 Print this Article Print this Article |
 Pubmed Pubmed |
 PMC PMC |
 PubReader PubReader |
 Export to Citation Export to Citation |
 Email Alerts Email Alerts |
 Open Access Open Access |
Supplementary
Share this article on :



Stats or Metrics
Article
Original Article
Exp Neurobiol 2020; 29(3): 189-206
Published online July 1, 2020
https://doi.org/10.5607/en20021
© The Korean Society for Brain and Neural Sciences
ERK Regulates NeuroD1-mediated Neurite Outgrowth via Proteasomal Degradation
Tae-young Lee1,2,3, In-Su Cho1, Narayan Bashyal1,2, Francisco J Naya4, Ming-Jer Tsai5, Jeong Seon Yoon1, Jung-Mi Choi1, Chang-Hwan Park6, Sung-Soo Kim1,2* and Haeyoung Suh-Kim1,2,3*
1Department of Anatomy, Ajou University School of Medicine, Suwon 16499, 2Department of Biomedical Sciences, Graduate School, Ajou University School of Medicine, Suwon 16499, 3Research Center, CelleBrain Ltd., Jeonju 54871, Korea, 4Department of Biology, Life Science and Engineering Building, Boston University, Boston, MA 00215, 5Department of Medicine and Program in Developmental Biology, Baylor College of Medicine, Houston, TX 77030, USA, 6Graduate School of Biomedical Science and Engineering, Hanyang University, Seoul 04763, Korea
Correspondence to: *To whom correspondence should be addressed.
Sung-Soo Kim, TEL: 82-31-219-5036, FAX: 82-31-219-5034
e-mail: kimdmg@ajou.ac.kr
Haeyoung Suh-Kim, TEL: 82-31-219-5036, FAX: 82-31-219-5039
e-mail: hysuh@ajou.ac.kr
This is an Open Access article distributed under the terms of the Creative Commons Attribution Non-Commercial License (http://creativecommons.org/licenses/by-nc/4.0) which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
Abstract
Neurogenic differentiation 1 (NeuroD1) is a class B basic helix-loop-helix (bHLH) transcription factor and regulates differentiation and survival of neuronal and endocrine cells by means of several protein kinases, including extracellular signal-regulated kinase (ERK). However, the effect of phosphorylation on the functions of NeuroD1 by ERK has sparked controversy based on context-dependent differences across diverse species and cell types. Here, we evidenced that ERK-dependent phosphorylation controlled the stability of NeuroD1 and consequently, regulated proneural activity in neuronal cells. A null mutation at the ERK-dependent phosphorylation site, S274A, increased the half-life of NeuroD1 by blocking its ubiquitin-dependent proteasomal degradation. The S274A mutation did not interfere with either the nuclear translocation of NeuroD1 or its heterodimerization with E47, its ubiquitous partner and class A bHLH transcription factor. However, the S274A mutant increased transactivation of the E-box-mediated gene and neurite outgrowth in F11 neuroblastoma cells, compared to the wild-type NeuroD1. Transcriptome and Gene Ontology enrichment analyses indicated that genes involved in axonogenesis and dendrite development were downregulated in NeuroD1 knockout (KO) mice. Overexpression of the S274A mutant salvaged neurite outgrowth in NeuroD1-deficient mice, whereas neurite outgrowth was minimal with S274D, a phosphomimicking mutant. Our data indicated that a longer protein half-life enhanced the overall activity of NeuroD1 in stimulating downstream genes and neuronal differentiation. We propose that blocking ubiquitin-dependent proteasomal degradation may serve as a strategy to promote neuronal activity by stimulating the expression of neuron-specific genes in differentiating neurons.
Graphical Abstract

Keywords: Neurogenic differentiation factor 1, Neurite outgrowth, Extracellular signal-regulated kinase, Phosphorylation
INTRODUCTION
Neurogenic differentiation factor 1 (NeuroD1) is a tissue-specific (class B) member of the basic helix-loop-helix (bHLH) protein family and plays a critical role in the commitment of neuronal precursors to neuronal differentiation [1].
NeuroD1 (also known as NeuroD and BETA2) is expressed in developing enteroendocrine cells of the pancreas and intestine and plays a critical role in the differentiation and survival of these cells [8,-10].
Extracellular signal-regulated kinases 1/2 (ERK1/2) appear to integrate long- and short-term nutrient sensing information in the nucleus of β-cells to maintain insulin homeostasis. The modulation of
In this study, we investigated the molecular mechanisms underlying the context-dependent activity of
MATERIALS AND METHODS
Animals
All experimental procedures using animals were approved by the Institutional Animal Care and Use Committee (IACUC) of Ajou University School of Medicine, South Korea (ethics number: 2015-0043).
Cell culture and transfection
HEK 293T and F11 cells were maintained in Dulbecco's modified Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum (FBS), 100 units/ml penicillin, and 100 µg/ml streptomycin and incubated in a humidified 5% CO2 incubator at 37°C. Cell lines with a maximum of ten passages were used. Cells were transiently transfected using polyethyleneimine (PEI; Polysciences, 23966) as a DNA carrier. To measure the half-life of
Preparation of lentiviral vectors
Lentiviral transfer vectors, encoding the Flag-tagged WT and mutant
Subcellular fractionation and western blotting analysis
HEK 293T cells cells (1×106 cells in a 100-mm2 dish) were transfected with
Cycloheximide (CHX) chase assay
HEK 293T cells were seeded in Dulbecco's modified Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum (FBS), 100 units/ml penicillin, and 100 µg/ml streptomycin at a density of 105 cells per well of a 6-well plate. One day later, PEI was used to transiently transfect cells with 3 µg of each plasmid encoding the WT and mutant
Ubiquitination assay
HEK 293T cells (105 cells per well of a 6-well plate) were transfected with 8 µg of each plasmid encoding the Myc-tagged WT (c-Myc-tag method was previously described [19]) or
Luciferase assay
One day prior to transfection, F11, P19, INS-1, and HeLa cells were seeded at a density of 1×105 cells per well of a 6-well plate. Transfection was carried out with 1 µg of each reporter plasmid of pGL3-NeuroD1, containing the 2.2 kb-long flanking sequences of the translation initiation site of
Neurite outgrowth analysis
Cultures were prepared from mouse primary cortical neurons at embryonic day 16.5 (E16.5). Briefly, the embryonic brains of
To assess neurite outgrowth, F11 cells were plated in growth medium (DMEM with 10% FBS, 100 units/ml penicillin, and 100 µg/ml streptomycin) and transiently co-transfected with lentiviral plasmids encoding the WT or S274 mutants, together with GFP, using PEI. Twenty-four hours later, transfected cells were re-plated into 6-well plates in a differentiation medium (DMEM with 0.5% (v/v) FBS, 100 units/ml penicillin, and 100 µg/ml streptomycin). Neurite outgrowth was monitored daily for up to 4 d [22]. Fluorescent live cell images were captured without immunostaining and analyzed using the NeuronJ plugin (version 1.4.3; Biomedical Imaging Group, Rotterdam, Netherlands) for ImageJ software (version 1.52; NIH, Bethesda, MD, USA). Data are presented in terms of the 25th, 50th, and 75th percentiles and standard error (S.E.) of more than 200 cells.
RNA sequencing analysis
Three biological replicates of
Quantitative RT-PCR
Total RNA was extracted from cultured cells or E18 mouse forebrain using the Hybrid-RTM RNA extraction kit (GeneAll, 305-101) and reverse-transcribed with SuperScriptTM III First-Strand Synthesis System (Invitrogen, 18080051). The 1st cDNA was amplified by polymerase chain reaction (PCR) using
Statistical analysis
All data are presented as mean±standard error (S.E.). All statistical analyses were performed using SigmaPlotTM v14 software (Systat Software, Inc., San Jose, CA, USA). One-way ANOVA and Holm-Sidak or Dunn’s tests were used, when deemed appropriate, to compare differences between the various groups. When necessary, the t-test and Mann-Whitney U test were also performed. To assess the normality of data, the Shapiro–Wilk test (p<0.05) was used.
RESULTS
Deletion of NeuroD1 reduced the neurite developmental genes
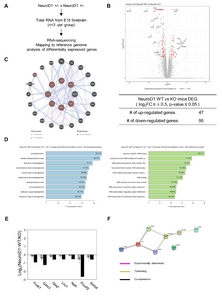
To investigate the role of NeuroD1 in differentiated neurons, we assessed the RNA profiles at gestational age 18d (E18), the latest period to consistently obtain the NeuroD1 knockout (KO) brains. RNA was extracted from the forebrains, including regions of the neocortex and hippocampus, of E18 NeuroD1 KO and WT littermates (Fig. 1A). Differentially expressed genes between KO and WT mice are represented as red dots and labelled in the volcano plot. A total of 47 genes were upregulated and 95 genes were downregulated (Log2 Fold Change (FC) cutoff value ≥0.5 and p-value ≤0.05) (Fig. 1B). A detailed gene list is provided in
Gene Ontology (GO) enrichment analysis revealed that downregulated genes were enriched in biological processes and molecular functions for cellular development and promoter binding/transcription, respectively (Fig. 1D). Since NeuroD1 is exclusively expressed in the nervous tissue during embryonic development, the data suggests that NeuroD1 deletion impaired neuronal transcription activity and neuronal development/differentiation. We further analyzed the relationship among the 95 downregulated genes using GeneMANIA web software [24] to delineate NeuroD1 functions specific for axon and telencephalic development. Seven genes, known to be co-expressed and co-localized with NeuroD1 (Fig. 1C), were detected, namely Gata3, Gbx2, Foxb1, Lhx1, Nefh, Pou4f2, and Slitrk6. The downregulation of these genes, triggered by NeuroD1 deletion, was further validated by quantitative reverse transcription polymerase chain reaction (qRT-PCR) analysis of RNA isolated from the embryonic forebrain using (Fig. 1E). Protein network analysis using STRING database revealed that six of the abovementioned proteins, excluding Slitrk6, interacted with NeuroD1 [25] (Fig. 1F). Gata3, Gbx2, Foxb1, Lhx1, and Pou4f2 are transcription factors that regulate neuronal differentiation in a diverse region of the brain during development and promote neurite outgrowth. Nefh and Slitrk6 are structural protein that regulates axonal elongation and stabilization (see the
Deletion of NeuroD1 reduces neurite outgrowth
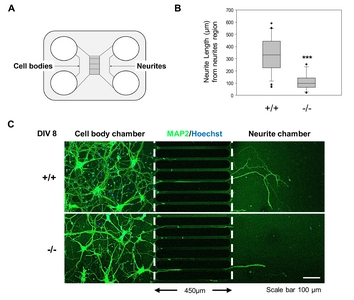
The neurite outgrowth in KO and WT primarily cultured neurons was assessed to investigate the role of NeuroD1 in differentiated neurons. An equal number of primary cortical neurons prepared from the forebrains of KO and WT embryos at E16.5 was plated in the cell body compartment of microfluidic chambers. At the 8th day
ERK regulates NeuroD1 protein stability
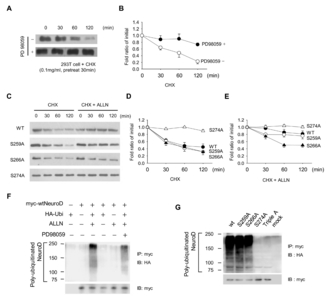
The effects of ERK on the functioning of NeuroD1 are distinct for insulinoma cell lines and
Following ubiquitination assay, multiple ubiquitin chains were identified in NeuroD1-containing precipitates, which were partially diminished by PD98059 (Fig. 3F). Importantly, the polyubiquitin chain entirely disappeared in the S274A mutant, whereas some chains remained in S259A or S266A mutants (Fig. 3G). These results indicated that NeuroD1 is a target of ubiquitin-proteasome-dependent degradation induced by ERK-dependent phosphorylation at S274.
NeuroD1 nuclear localization is reduced in the S274D mutant
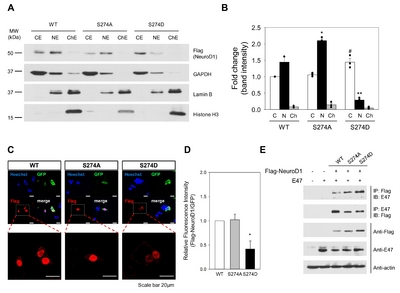
A previous study showed that S274 mutation affects nuclear localization in insulinoma cell lines [18]. We tested whether the effects of ERK-dependent phosphorylation are reproduced equally in cells other than those of insulinoma cell lines. We transfected HEK 293T cells with expression vectors for WT and mutants, and the cell lysate was fractionated to isolate three compartments: cytosolic, nuclear, and chromatin-bound extracts. Subcellular fractionation methods were validated by the presence of GAPDH (cytosol), Lamin B (nucleus), and Histone H3 (chromatin-bound) (Fig. 4A). The WT and S274A mutant were expressed in both the cytosol and nucleus, which were expressed at higher levels in the nucleus. This tendency was more pronounced with regards to the S274A mutant (Fig. 4B). In contrast, S274D is mostly expressed in the cytosol but barely present in the nuclear fraction. Immunostaining with an anti-Flag antibody revealed strong signals for WT and S274A in the nucleus, but weak signals for S274D (Fig. 4C and D). These results suggest that the nuclear translocation of NeuroD1 is retarded following ERK-phosphorylation.
To determine whether S274 mutations alter the ability of NeuroD1 proteins to heterodimerize with class A protein families, such as E47, coimmunoprecipitation assays were performed. HEK 293T cells were co-transfected with expression vectors for the WT and mutant NeuroD1, together with E47. Since both NeuroD1 and E47 independently contain nuclear localization signals and form heterodimers in the nucleus, we used cytosolic fractions for co-immunoprecipitation. The immunoprecipitates containing Flag-tagged WT, S274A, and S274D were immunoblotted to detect the presence of E47. The immunoprecipitate content of E47 was proportional to the amount of total immune-complex in the cytosol. This suggests that S274 mutations do not significantly alter the heterodimer-binding activity of NeuroD1 and E47 in the cytosol (Fig. 4E). By comparison, the NeuroD1 content in E47-containing immunoprecipitates appeared slightly higher in case of the WT than that in case of the mutants. It is not practically feasible to delineate the underlying mechanisms for this at present, as several factors may influence the coimmunoprecipitation efficiency, including relative protein amounts, E47 homodimer formation, and steric hindrance caused by other protein interactions. Importantly, S274 phosphorylation, as demonstrated by S274D, does not significantly alter the ability of NeuroD1 to form heterodimers with E47.
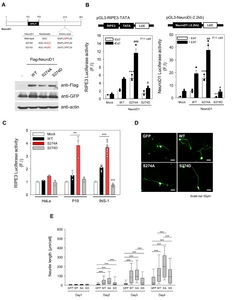
S274 phosphorylation attenuates transactivation potential of NeuroD1
We tested whether ERK-induced proteolysis correlated with the ability of NeuroD1 to transactivate downstream genes in neuronal cells. Previous studies evidenced ERK transactivated the C-terminus of NeuroD1 (155-355 aa) that was conjugated to the GAL4 DNA-binding domain in insulinoma cell lines, such as MIN6, βTC3, or INS-1 [17, 18]. Unlike previous studies, we used full length (1-355 aa) NeuroD1 proteins in the neuronal cell line, F11. For this, we generated pLenti (lentivirus transfer) vectors for concomitant expression of the WT or S274 mutants together with GFP protein. Concomitant expression of GFP and Flag-tagged NeuroD1 proteins was validated by western blot analysis (Fig. 5A). Since class B bHLH transcription factors, such as NeuroD1, form heterodimers with their class A partners, such as E47 [26], transfection was performed with and without E47. A reporter gene containing rat insulin promoter element 3 (RIPE3), an E-box containing enhancer that is known to be activated by NeuroD1, was used [9, 12]. In contrast to that of insulinoma cell lines, the S274A mutant activated RIPE3, containing the reporter gene, by 5-fold, which further increased to 11-fold when transfection was performed with E47. In contrast, S274D, a non-phosphorylated mutant, only activated RIPE3 by 2-fold, even when transfected with E47. To further verify the NeuroD’s functions, a reporter gene containing the 2.2-kb promoter region of NeuroD1 [9] was used as it was evidenced to be autoregulated by NeuroD1 through E-box sequences [27]. The 2.2 kb fragment of NeuroD1 promoter was also activated by S274A by 40- and 18-fold with and without E47, respectively (Fig. 5B). NeuroD1-mediated gene activation is cell-type specific. S274A enhanced the expression of both reporter genes in pluripotent P19 cells, or INS-1 insulinoma cells, and in F11 cells but not in HeLa cells (Fig. 5C). Importantly, the full length NeuroD1 can transactivate the E-box containing promoter in both F11 neuronal and INS-1 insulinoma cells. These results clearly indicate that ERK-dependent phosphorylation at S274 exerts negative effects on the NeuroD1 protein.
We used F11 cells to investigate whether increased NeuroD1 activity promotes neurite outgrowth in differentiated neurons. F11 cells provide an
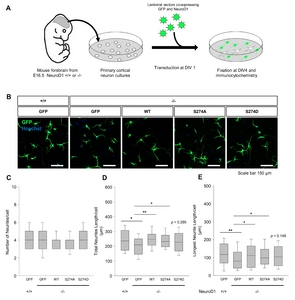
WT and S274A rescue neurite outgrowth in NeuroD1 KO neurons
To determine whether the stimulatory effect of NeuroD1 observed in F11 neuronal cells is also conserved in primary neurons, rescue experiments were performed with the WT and mutants from primary cortical neurons isolated from NeuroD1 KO embryos. On day 4, total and longest neurite lengths were reduced in KO neurons compared to those of the WT, indicating that our approach of using a 4-day culture was as valid as the 8-day long microfluidic chamber assay in terms of dissociated cultures. Several types of lentiviral vectors, encoding GFP in conjunction with WT or mutant NeuroD1 proteins, were transduced to KO neurons to assess the effect of ERK-dependent phosphorylation (Fig. 6A~C). Expression of the WT rescued neurite outgrowth in KO neurons, which was similar to the case for WT neurons (p<0.01). Importantly, the S274A mutant also significantly enhanced total and longest neurite lengths similar to that of WT neurons (p<0.05). By comparison, S274D expression could increase neurite length to a lesser degree; thus, the difference between S274D and the GFP control was statistically insignificant (p=0.295). The number of neurites was around 4 per cell in all experimental groups (Fig. 6D~G). The results suggest that ERK-dependent phosphorylation of S274 may attenuate the function of NeuroD1 in neurite extension without affecting the formation of new neurites.
DISCUSSION
NeuroD1 is a tissue-specific transcription factor and its functions are regulated by several kinases that transmit the growth- and activity-related extracellular signals to the nucleus. Several studies conducted over the past decade have evidenced that transcriptional activity of NeuroD1 appears to be oppositely regulated by ERK in neurons and pancreatic β cells. In insulinoma cell lines, ERK, triggered by glucose stimulation, appeared to activate the transactivation potential of NeuroD1, when functioning as a hybrid protein with the GAL4 DNA-binding domain. In contrast, a phosphorylation-null mutation at the ERK phosphorylation site increased proneural functions of NeuroD1 in early embryogenesis. The present study has clarified a long-standing enigma regarding the impact of ERK phosphorylation on functions of NeuroD1.
Like other members of the class B bHLH transcription factor family [28, 29], the NeuroD1 protein undergoes rapid decay via ubiquitin-dependent degradation (Fig. 3). The degradation rate declines when the ERK pathway is blocked with a MEK inhibitor. Ala substitution at the ERK phosphorylation site (S274A) also lowers the protein decay rate and increases the amount of protein in the nuclear compartment (Fig. 4). Unlike previous studies [17, 18] using a hybrid of NeuroD1 (156-355) with Gal4 DNA-binding domain (DBD), the current study evidenced that full-length NeuroD1 proteins are capable of upregulating the expression of downstream reporter genes and that the stimulatory effect is higher with the S274A mutation compared to the WT (Fig. 5). Similar results were obtained with full-length NeuroD1 in P19, an embryonic carcinoma cell line, and INS-1, a rat insulinoma cell line (Fig. 5C), suggesting that the ERK pathway negatively regulates the functions of NeuroD1. We summarized of the effect S274A mutation in diverse cell types in Table 2. Likewise, neurite outgrowth is accelerated with the S274A mutation than that of the WT in F11 neuronal cells (Fig. 5). The S274A mutation does not alter the innate activity of the NeuroD1 protein with regards to nuclear localization or heterodimer formation with E47 (Fig. 4). Thus, the increased activity of S274A can be explained by its high content, due to reduced decay rates or increased protein stability.
In contrast, S274D, a phosphomimetic mutant of ERK, demonstrated poor activity in transactivating reporter genes or inducing neurite outgrowth compared to that of the WT protein. Although the cytosolic content of S274D was relatively high compared to that of the WT or S274A, the nuclear localization of S274D was significantly attenuated (Fig. 4A~D). Thus, the poor activity of S274D in transactivation or neurite promotion is due to the low content of functional complexes containing the S274D protein in the nuclear compartment, and consequently low occupancy of the promoter. E47 contains nuclear localization sequences at 165-169 aa and 539-554 aa; thus, it is able to direct nuclear localization of a partner protein in heterodimer complexes containing a tissue-restricted class B bHLH protein partner, such as MyoD [28]. Since S274D in the cytosolic compartment can interact and form a heterodimer with E47 (Fig. 4D), the low content of S274D in the nuclear compartment implies that the S274D-containing complex is prohibited from the nuclear compartment, which cannot be negated by the nuclear localization sequences of E47. Further studies are necessary to reveal whether and, if so, how ERK-dependent phosphorylation induces tethering of the heterodimer complex to cytosolic adaptor(s).
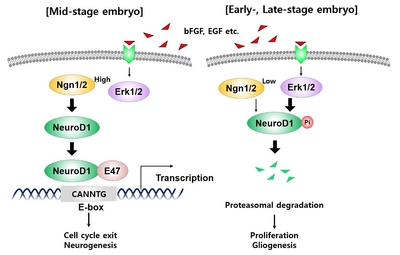
Although this study has revealed the molecular mechanism of Erk-dependent NeuroD1 regulation via observing neurite outgrowth in differentiated neuronal cells, this finding can be expanded to elaborate cell fate determination of the uncommitted stem cells. During the development of forebrain, ERK signals exert opposite effects on neuronal and astroglial differentiation. ERK suppresses neuronal differentiation by inhibiting the expression of Neurogenins 2 (Neurog2), a proneural transcription factor, in dorsal progenitor cells during cell cycle progression. At the commitment stages of progenitor cells, transcription of Neurog2 and NeuroD1 (the target gene of Neurog2) increases as ERK signals decline. As a result, the cells in the dorsal forebrain exit the cell cycle and terminally differentiate into glutamatergic neurons [30]. Indeed, NeuroD1 is known to activate a cyclin-dependent kinase inhibitor, p21 and thereby induce cell cycle exit [31]. Thus, the negative regulation of Neurog2 at the transcriptional level and NeuroD1 at the post-translational level by ERK shown in the present study may demonstrate potential mechanisms for prohibiting premature cell cycle exit while maintaining cortical progenitor pools in the mid-embryonic stages. After entering late embryonic stages, the dorsal progenitor cells acquire astroglial competence and generate astrocytes at the expense of neurons [32]. Suppression of RAS-ERK pathway by deletion of Mek1/2 (also known as MAPK/ERK kinase 1/2) or deletion of ERK1/2 induces depletion of astrocytes in postnatal brain [33, 34], suggesting that ERK plays a role in the astroglial lineage progression of dorsal progenitor cells. Our finding of the negative regulation of NeuroD1 protein by ERK may provide a clue to elucidate the previously unexplored mechanisms for driving the genetic switch of dorsal progenitor cells from neurogenic-to-gliogenic competence after Neurog2 expression declines in the late stage of development (Fig. 7) [35,-37]. Impairments in lineage progression caused by malfunction of RAS-ERK signaling lead to alterations in the cortical cytoarchitecture, which is thought to represent the major underlying cause of several neurological disorders, including microcephaly or megalocephaly, and more subtle neurodevelopmental diseases, including schizophrenia, autism, epilepsy, and RASopathies [38,-40].
Transcriptome analysis and GO enrichment analysis indicated that the genes involved in axonogenesis and dendrite development were downregulated in NeuroD1 KO mice (Fig. 1). In the primary neurons obtained from NeuroD1 KO embryos, neurite outgrowth is attenuated, suggesting that NeuroD1 plays a role in proneural activity regarding the development of neuronal processes. F11 cells are a hybridoma cell line between mouse neuroblastoma and rat dorsal root ganglion (pseudo-unipolar) cells and extend bipolar neurites in the presence of dibutyryl-cAMP. Without it, the F11 cells barely extend neurites up to day 3 (Fig. 5D and E), suggesting that innate neurogenic factors are limited in F11 cells. Overexpression of WT, S274D, and S274A NeuroD1 can promote neurite outgrowth in F11 cells with the highest magnitude for S274A and the lowest for S274D (Fig. 5). The data are consistent with the proteins’ half-lives (Fig. 3) and transactivation potential of downstream reporter genes (Fig. 5B).
By comparison, the effect of S274A is not prominent compared to the WT in NeuroD1 -/- primary neurons (Fig. 6). It can be partly due to that axonal and dendritic morphogenesis in cortical neurons is regulated by pleiotropic factors and the loss of NeuroD1 can be compensated by other bHLH proteins, such as NeuroD2, MATH2, MATH3, and Atoh7 as shown in NeuroD1 knockout mice [5, 41, 42]. Thus, NeuroD1 -/- cortical neurons can extend neurites, although to a lesser degree, compared to WT neurons (Fig. 1 and 6). Therefore, the influence of S274A may be minimal in the rescue experiments using NeuroD1 -/- cortical neurons compared to the one in F11 cells. Importantly, S274D is less effective to rescue the NeuroD1’s functions (Fig. 6), supporting our hypothesis that phosphorylation of S274 is important to determine the neuritogenic function of NeuroD1.
A previous study evidenced that NeuroD1 can be phosphorylated by an activity-induced protein kinase, CaMKII, at S336 and the S336A mutation destroyed the ability of NeuroD1 to stimulate dendritic growth in Bcl2-overexpressed cerebellar granule neurons [43, 44]. However, similar to that of insulinoma cells [17], the transactivation activity of NeuroD1 is not altered by S336 mutations when tested with chimeric proteins fused to the Gal4 DNA-binding domain [44]. Further studies using the full length NeuroD1 protein may be helpful in revealing how NeuroD1 phosphorylation, triggered by CaMKII, upregulates dendritic growth in mature neurons.
The RNA sequencing analysis of 95 downregulated genes revealed the functions of NeuroD1 with respect to the GO terms of promoter binding, RNA polymerase II, and cellular developmental processes (Fig. 1D). Subsequent analysis using GeneMANIA revealed seven core genes related to the GO term of neurite outgrowth (Fig. 1). Gata3, Gbx2, Foxb1, Lhx1, and Pou4f2 are transcription factors that regulate neuronal differentiation in a diverse region of the brain during development and promote neurite outgrowth [45,-49]. Nefh (Neurofilament, Heavy polypeptide) is a cytoskeletal element of axons and Slitrk6 (SLIT and NTRK like family member 6) is an integral membrane protein, both of which regulates axonal elongation and stabilization [50, 51]. Interestingly, all these genes contain potential E-box sequences within 1 kb upstream of the transcription start site (
Recently, NeuroD1 has emerged as an important genetic therapeutic strategy to treat the diseased brain where adult neurogenesis or synaptic integration of newborn neurons are hampered. In Alzheimer’s disease model (5xFAD), overexpression of NeuroD1 using a retroviral vector can induce reprogramming of reactive astroglial cells into functional neurons [55]. Similarly, overexpression of NeuroD1 also can rescue spatial memory in aged AD mice (APPxPS1) by accelerating neuronal maturation of hippocampal progenitor cells and promote functional integration of newborn neurons [56, 57]. S274A has a longer protein half-life than WT NeuroD1, making it a better choice for overexpressing NeuroD1 to alleviate neuronal loss in a diseased brain. By blocking the ubiquitin-dependent degradation of endogenous NeuroD1 protein may provide an alternative therapeutic strategy that promotes neuronal activity by enhancing the expression of NeuroD1’s target genes.
ACKNOWLEDGEMENTS
This study was supported by grants from the National Research Foundation of Korea (NRF-2018M3A9G1082594- to HS-K.) and the INNOPOLIS Foundation of Jeonbuk (2019-JB-RD-0048-01101). We would like to thank Dr. Jacqueline E. Lee (Dep. Molecular, Cellular & Developmental Biology, University of Colorado, Boulder, USA) and Dr. Kang Ho Chul (Dep. Physiol. Ajou University, Suwon, Korea) for providing Myc-tagged
Figures
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Tables
qRT-PCR primer list
| Genes | Forward primer sequence 5’-3’ | Reverse primer sequence 3’-5’ |
|---|---|---|
| Foxb1 | CTTCAAGGTGCTCAAGTCAGAC | GTTCTCGATAGCAAAGGGATGC |
| Gata3 | CTCGGCCATTCGTACATGGAA | GGATACCTCTGCACCGTAGC |
| Gbx2 | CAACTTCGACAAAGCCGAGG | ACTCGTCTTTCCCTTGCCCT |
| Lhx1 | CCCATCCTGGACCGTTTCC | CGCTTGGAGAGATGCCCTG |
| Nefh | AGACCCCCGTCAAGGAAGG | CTTCTCAGGGGATTTCGCCT |
| Pou4f2 | TGGACATCGTCTCCCAGAGTA | GTGTTCATGGTGTGGTAAGTGG |
| Slitrk6 | AGGCTCTTGCGACACTCTTTG | GTGGCACACTGATTTGGGATAAT |
Summary of the effect of S274A mutation in diverse cell types
| Cell type | Cell name | NeuroD1 construct | Protein length (amino acid) | Localization | Transcriptional analysis | References | |
|---|---|---|---|---|---|---|---|
| Activity | Methods | ||||||
| Pancrease | MIN6 | Gal4-DBD-NeuroD1 | 156~355 | Cytosol>Nuclei | Low | CAT | [18] |
| βTC3 | Gal4-DBD-NeuroD1 | 156~355 | n.a. | Low | Luciferase | [17] | |
| INS-1 | Gal4-DBD-NeuroD1 | 156~355 | n.a. | Low | Luciferase | [17] | |
| INS-1 | Flag-NeuroD1 | 1~355 | n.a. | High | Luciferase | In this study | |
| Cervical cancer | HeLa | Flag-NeuroD1 | 1~355 | n.a. | High | Luciferase | In this study |
| Kidney | 293T | Flag-NeuroD1 | 1~355 | Cytosol | High | Luciferase | In this study |
| Neuroblastoma | P19 | Flag-NeuroD1 | 1~355 | n.a. | High | Luciferase | In this study |
| F11 | Flag-NeuroD1 | 1~355 | n.a. | High | Luciferase | In this study | |
DBD, DNA Binding Domain.
References
- Dennis DJ, Han S, Schuurmans C (2019) bHLH transcription factors in neural development, disease, and reprogramming. Brain Res 1705: 48-65


- Lee JE, Hollenberg SM, Snider L, Turner DL, Lipnick N, Weintraub H (1995) Conversion of
Xenopus ectoderm into neurons by NeuroD, a basic helix-loop-helix protein. Science 268: 836-844
- Kim WY, Fritzsch B, Serls A, Bakel LA, Huang EJ, Reichardt LF, Barth DS, Lee JE (2001) NeuroD-null mice are deaf due to a severe loss of the inner ear sensory neurons during development. Development 128: 417-426

- Liu M, Pereira FA, Price SD, Chu MJ, Shope C, Himes D, Eatock RA, Brownell WE, Lysakowski A, Tsai MJ (2000) Essential role of BETA2/NeuroD1 in development of the vestibular and auditory systems. Genes Dev 14: 2839-2854
- Miyata T, Maeda T, Lee JE (1999) NeuroD is required for differentiation of the granule cells in the cerebellum and hippocampus. Genes Dev 13: 1647-1652
- Schwab MH, Bartholomae A, Heimrich B, Feldmeyer D, Druffel-Augustin S, Goebbels S, Naya FJ, Zhao S, Frotscher M, Tsai MJ, Nave KA (2000) Neuronal basic helix-loop-helix proteins (NEX and BETA2/Neuro D) regulate terminal granule cell differentiation in the hippocampus. J Neurosci 20: 3714-3724
- Gao Z, Ure K, Ables JL, Lagace DC, Nave KA, Goebbels S, Eisch AJ, Hsieh J (2009) Neurod1 is essential for the survival and maturation of adult-born neurons. Nat Neurosci 12: 1090-1092
- Naya FJ, Huang HP, Qiu Y, Mutoh H, DeMayo FJ, Leiter AB, Tsai MJ (1997) Diabetes, defective pancreatic morphogenesis, and abnormal enteroendocrine differentiation in BETA2/neuroD-deficient mice. Genes Dev 11: 2323-2334
- Huang HP, Liu M, El-Hodiri HM, Chu K, Jamrich M, Tsai MJ (2000) Regulation of the pancreatic islet-specific gene BETA2 (neuroD) by neurogenin 3. Mol Cell Biol 20: 3292-3307
- Schonhoff SE, Giel-Moloney M, Leiter AB (2004) Minireview: development and differentiation of gut endocrine cells. Endocrinology 145: 2639-2644
- Mutoh H, Fung BP, Naya FJ, Tsai MJ, Nishitani J, Leiter AB (1997) The basic helix-loop-helix transcription factor BETA2/NeuroD is expressed in mammalian enteroendocrine cells and activates secretin gene expression. Proc Natl Acad Sci U S A 94: 3560-3564
- Naya FJ, Stellrecht CM, Tsai MJ (1995) Tissue-specific regulation of the insulin gene by a novel basic helix-loop-helix transcription factor. Genes Dev 9: 1009-1019
- Luo H, Chen R, Yang R, Liu Y, Chen Y, Shu Y, Chen H (2014) Reprogramming of mice primary hepatocytes into insulin-producing cells by transfection with multicistronic vectors. J Diabetes Res 2014: 716163
- Lee SH, Rhee M, Kim JW, Yoon KH (2017) Generation of insulin-expressing cells in mouse small intestine by Pdx1, MafA, and BETA2/NeuroD. Diabetes Metab J 41: 405-416
- Ham DS, Shin J, Kim JW, Park HS, Cho JH, Yoon KH (2013) Generation of functional insulin-producing cells from neonatal porcine liver-derived cells by PDX1/VP16, BETA2/NeuroD and MafA. PLoS One 8: e79076
- Song YD, Lee EJ, Yashar P, Pfaff LE, Kim SY, Jameson JL (2007) Islet cell differentiation in liver by combinatorial expression of transcription factors neurogenin-3, BETA2, and RIPE3b1. Biochem Biophys Res Commun 354: 334-339
- Khoo S, Griffen SC, Xia Y, Baer RJ, German MS, Cobb MH (2003) Regulation of insulin gene transcription by ERK1 and ERK2 in pancreatic beta cells. J Biol Chem 278: 32969-32977
- Petersen HV, Jensen JN, Stein R, Serup P (2002) Glucose induced MAPK signalling influences NeuroD1-mediated activation and nuclear localization. FEBS Lett 528: 241-245
- Dufton C, Marcora E, Chae JH, McCullough J, Eby J, Hausburg M, Stein GH, Khoo S, Cobb MH, Lee JE (2005) Context-dependent regulation of NeuroD activity and protein accumulation. Mol Cell Neurosci 28: 727-736
- Jiang W, Hua R, Wei M, Li C, Qiu Z, Yang X, Zhang C (2015) An optimized method for high-titer lentivirus preparations without ultracentrifugation. Sci Rep 5: 13875
- Hwung YP, Gu YZ, Tsai MJ (1990) Cooperativity of sequence elements mediates tissue specificity of the rat insulin II gene. Mol Cell Biol 10: 1784-1788
- Cho JH, Kwon IS, Kim S, Ghil SH, Tsai MJ, Kim YS, Lee YD, Suh-Kim H (2001) Overexpression of BETA2/NeuroD induces neurite outgrowth in F11 neuroblastoma cells. J Neurochem 77: 103-109
- Reimand J, Arak T, Adler P, Kolberg L, Reisberg S, Peterson H, Vilo J (2016) g:Profiler-a web server for functional interpretation of gene lists (2016 update). Nucleic Acids Res 44: W83-W89
- Montojo J, Zuberi K, Rodriguez H, Kazi F, Wright G, Donaldson SL, Morris Q, Bader GD (2010) GeneMANIA Cytoscape plugin: fast gene function predictions on the desktop. Bioinformatics 26: 2927-2928
- Szklarczyk D, Gable AL, Lyon D, Junge A, Wyder S, Huerta-Cepas J, Simonovic M, Doncheva NT, Morris JH, Bork P, Jensen LJ, Mering CV (2019) STRING v11: protein-protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res 47: D607-D613
- Glick E, Leshkowitz D, Walker MD (2000) Transcription factor BETA2 acts cooperatively with E2A and PDX1 to activate the insulin gene promoter. J Biol Chem 275: 2199-2204
- Fang HB, Mi Y, Zhang Y, Wu NH, Shen YF (2010) HDAC3 augments the autoregulation of neuroD gene in P19 cells. Neuroreport 21: 19-23
- Lingbeck JM, Trausch-Azar JS, Ciechanover A, Schwartz AL (2005) E12 and E47 modulate cellular localization and proteasome-mediated degradation of MyoD and Id1. Oncogene 24: 6376-6384
- Hindley C, Ali F, McDowell G, Cheng K, Jones A, Guillemot F, Philpott A (2012) Post-translational modification of Ngn2 differentially affects transcription of distinct targets to regulate the balance between progenitor maintenance and differentiation. Development 139: 1718-1723
- Li S, Mattar P, Dixit R, Lawn SO, Wilkinson G, Kinch C, Eisenstat D, Kurrasch DM, Chan JA, Schuurmans C (2014) RAS/ERK signaling controls proneural genetic programs in cortical development and gliomagenesis. J Neurosci 34: 2169-2190
- Mutoh H, Naya FJ, Tsai MJ, Leiter AB (1998) The basic helix-loop-helix protein BETA2 interacts with p300 to coordinate differentiation of secretin-expressing enteroendocrine cells. Genes Dev 12: 820-830
- Hirabayashi Y, Itoh Y, Tabata H, Nakajima K, Akiyama T, Masuyama N, Gotoh Y (2004) The Wnt/beta-catenin pathway directs neuronal differentiation of cortical neural precursor cells. Development 131: 2791-2801
- Li X, Newbern JM, Wu Y, Morgan-Smith M, Zhong J, Charron J, Snider WD (2012) MEK is a key regulator of gliogenesis in the developing brain. Neuron 75: 1035-1050
- Imamura O, Pagès G, Pouysségur J, Endo S, Takishima K (2010) ERK1 and ERK2 are required for radial glial maintenance and cortical lamination. Genes Cells 15: 1072-1088
- Tabata H (2015) Diverse subtypes of astrocytes and their development during corticogenesis. Front Neurosci 9: 114
- Kriegstein A, Alvarez-Buylla A (2009) The glial nature of embryonic and adult neural stem cells. Annu Rev Neurosci 32: 149-184
- Beattie R, Hippenmeyer S (2017) Mechanisms of radial glia progenitor cell lineage progression. FEBS Lett 591: 3993-4008
- Kang M, Lee YS (2019) The impact of RASopathy-associated mutations on CNS development in mice and humans. Mol Brain 12: 96
- Silbereis JC, Pochareddy S, Zhu Y, Li M, Sestan N (2016) The cellular and molecular landscapes of the developing human central nervous system. Neuron 89: 248-268
- Desikan RS, Barkovich AJ (2016) Malformations of cortical development. Ann Neurol 80: 797-810
- Cho JH, Tsai MJ (2006) Preferential posterior cerebellum defect in BETA2/NeuroD1 knockout mice is the result of differential expression of BETA2/NeuroD1 along anterior-posterior axis. Dev Biol 290: 125-138
- Mao CA, Cho JH, Wang J, Gao Z, Pan P, Tsai WW, Frishman LJ, Klein WH (2013) Reprogramming amacrine and photoreceptor progenitors into retinal ganglion cells by replacing
NeuroD1 with Atoh7. Development 140: 541-551
- Lee YY, Choi HJ, Lee SY, Park SY, Kang MJ, Han J, Han JS (2020) Bcl-2 overexpression induces neurite outgrowth via the Bmp4/Tbx3/NeuroD1 cascade in H19-7 cells. Cell Mol Neurobiol 40: 153-166
- Gaudillière B, Konishi Y, de la Iglesia N, Yao Gl, Bonni A (2004) A CaMKII-NeuroD signaling pathway specifies dendritic morphogenesis. Neuron 41: 229-241
- Appler JM, Lu CC, Druckenbrod NR, Yu WM, Koundakjian EJ, Goodrich LV (2013) Gata3 is a critical regulator of cochlear wiring. Version 2. J Neurosci 33: 3679-3691
- Chen L, Chatterjee M, Li JY (2010) The mouse homeobox gene Gbx2 is required for the development of cholinergic interneurons in the striatum. Version 2. J Neurosci 30: 14824-14834
- Lui NC, Tam WY, Gao C, Huang JD, Wang CC, Jiang L, Yung WH, Kwan KM (2017) Lhx1/5 control dendritogenesis and spine morphogenesis of Purkinje cells via regulation of Espin. Nat Commun 8: 15079
- Wang SW, Mu X, Bowers WJ, Kim DS, Plas DJ, Crair MC, Federoff HJ, Gan L, Klein WH (2002) Brn3b/Brn3c double knockout mice reveal an unsuspected role for Brn3c in retinal ganglion cell axon outgrowth. Development 129: 467-477
- Alvarez-Bolado G, Zhou X, Voss AK, Thomas T, Gruss P (2000) Winged helix transcription factor Foxb1 is essential for access of mammillothalamic axons to the thalamus. Development 127: 1029-1038
- Lee S, Shea TB (2014) The high molecular weight neurofilament subunit plays an essential role in axonal outgrowth and stabilization. Biol Open 3: 974-981
- Katayama K, Zine A, Ota M, Matsumoto Y, Inoue T, Fritzsch B, Aruga J (2009) Disorganized innervation and neuronal loss in the inner ear of Slitrk6-deficient mice. PLoS One 4: e7786
- Seo S, Lim JW, Yellajoshyula D, Chang LW, Kroll KL (2007) Neurogenin and NeuroD direct transcriptional targets and their regulatory enhancers. EMBO J 26: 5093-5108
- Lin Z, Cantos R, Patente M, Wu DK (2005) Gbx2 is required for the morphogenesis of the mouse inner ear: a downstream candidate of hindbrain signaling. Development 132: 2309-2318
- Chatterjee S, Kraus P, Lufkin T (2010) A symphony of inner ear developmental control genes. BMC Genet 11: 68
- Guo Z, Zhang L, Wu Z, Chen Y, Wang F, Chen G (2014) In vivo direct reprogramming of reactive glial cells into functional neurons after brain injury and in an Alzheimer's disease model. Cell Stem Cell 14: 188-202
- Richetin K, Leclerc C, Toni N, Gallopin T, Pech S, Roybon L, Rampon C (2015) Genetic manipulation of adult-born hippocampal neurons rescues memory in a mouse model of Alzheimer's disease. Brain 138(Pt 2): 440-455
- Richetin K, Moulis M, Millet A, Arràzola MS, Andraini T, Hua J, Davezac N, Roybon L, Belenguer P, Miquel MC, Rampon C (2017) Amplifying mitochondrial function rescues adult neurogenesis in a mouse model of Alzheimer's disease. Neurobiol Dis 102: 113-124