Articles

Article Tools
 View Full Text View Full Text |
 Abstract Abstract |
 Article as PDF Article as PDF |
 Print this Article Print this Article |
 Pubmed Pubmed |
 PMC PMC |
 PubReader PubReader |
 Export to Citation Export to Citation |
 Email Alerts Email Alerts |
 Open Access Open Access |
Share this article on :



Stats or Metrics
Article
Original Article
Exp Neurobiol 2019; 28(6): 670-678
Published online December 31, 2019
https://doi.org/10.5607/en.2019.28.6.670
© The Korean Society for Brain and Neural Sciences
The Molecular Signatures of Acute-immobilization-induced Antinociception and Chronic-immobilization-induced Antinociceptive Tolerance
Jing-Hui Feng, Hee-Jung Lee and Hong-Won Suh*
Department of Pharmacology and Institute of Natural Medicine, College of Medicine, Hallym University, Chuncheon 24252, Korea
Correspondence to: *To whom correspondence should be addressed.
TEL: 82-33-248-2614, FAX: 82-33-248-2612
e-mail: hwsuh@hallym.ac.kr.
This is an Open Access article distributed under the terms of the Creative Commons Attribution Non-Commercial License(http://creativecommons.org/licenses/by-nc/4.0) which permits unrestricted non-commercial use, distribution, andreproduction in any medium, provided the original work is properly cited.
Abstract
In the present study, the productions of antinociception induced by acute and chronic immobilization stress were compared in several animal pain models. In the acute immobilization stress model (up to 1 hr immobilization), the antinociception was produced in writhing, tail-flick, and formalin-induced pain models. In chronic immobilization stress experiment, the mouse was enforced into immobilization for 1 hr/day for 3, 7, or 14 days, then analgesic tests were performed. The antinociceptive effect was gradually reduced after 3, 7 and 14 days of immobilization stress. To delineate the molecular mechanism involved in the antinociceptive tolerance development in the chronic stress model, the expressions of some signal molecules in dorsal root ganglia (DRG), spinal cord, hippocampus, and the hypothalamus were observed in acute and chronic immobilization models. The COX-2 in DRG, p-JNK, p-AMPKα1, and p-mTOR in the spinal cord, p-P38 in the hippocampus, and p-AMPKα1 in the hypothalamus were elevated in acute immobilization stress, but were reduced gradually after 3, 7 and 14 days of immobilization stress. Our results suggest that the chronic immobilization stress causes development of tolerance to the antinociception induced by acute immobilization stress. In addition, the COX-2 in DRG, p-JNK, p-AMPKα1, and p-mTOR in the spinal cord, p-P38 in the hippocampus, and p-AMPKα1 in the hypothalamus may play important roles in the regulation of antinociception induced by acute immobilization stress and the tolerance development induced by chronic immobilization stress.
Graphical Abstract
Keywords: Acute stress, Chronic stress, Antinociception, Tolerance, Signal molecule
INTRODUCTION
Stress can produce a series of physiological, neurological, and behavioral changes in the human body. Especially, stress elicits antinociceptive effects, of which the types include electric foot-shock, cold water swim, glucoprivation, forced swimming, defeat, and immobilization [1, 2]. Among those different types of stress, immobilization (IMO) stress is useful for studying stress-induced alteration of pain senses. A series of studies have shown that the acute IMO stress produces antinociception in selected animal models of pain, including the hot plate, tail-flick and formalin tests [3–5]. In addition, Pezalla and Dicig [6] reported that the acute IMO stress alters the antinociceptive effect in frogs. Although the production of the antinociception induced by acute IMO stress has been reported, the effect of chronic IMO stress on acute IMO stress-induced antinociception has not been well characterized.
Numerous studies have demonstrated that stress is related to the activation of multiple signaling pathways, such as MAPK-, COX-, iNOS- signaling molecules. Yang et al. [7] reported that chronic restraint stress induces MAPK phosphorylation. In addition, COX-2 is also activated by stress [8–10]. Moreover, oxidative stress elevated the expression of iNOS [11–13]. On the other hand, a series of studies have suggested that there are several molecules are related to antinociception. AMPK has recently emerged as a novel target for the treatment of pain [14]. Moreover, ERK plays a vital role in the development of neuropathic pain [15]. Furthermore, mTOR was a new potential target for chronic pain, hyperalgesia, and neuropathic pain [16–19]. However, the exact roles of these signaling molecules in the regulation of IMO stress-induced antinociception have not been delineated yet.
Although several studies on acute IMO stress-induced antinociception and alterations of signal molecules located peripherally and centrally have been performed, the effects of chronic IMO stress on acute IMO stress-induced antinociception, as well as expression of signal molecules induced by acute IMO stress have not been well characterized. Thus, the present study was designed to examine the effect of acute and chronic IMO stress on pain behavior in various pain models. Furthermore, the possible roles of signal molecules such as AMPKα1, JNK, p-38, COX2, mTOR proteins in the production antinociception by acute IMO stress and the development of antinociceptive tolerance by chronic IMO stress were investigated.
MATERIALS AND METHODS
These experiments were approved by the University of Hallym Animal Care and Use Committee (Registration Number: Hallym R1(2017–57). All procedures were conducted in accordance with the ‘Guide for Care and Use of Laboratory Animals’ published by the National Institutes of Health and the ethical guidelines of the International Association for the Study of Pain.
Experimental animals
Male ICR mice (MJ Co., Seoul, Korea) weighing 20~25 g were used for all the experiments. Animals were housed 5 per cage in a room maintained at 22±0.5°C with an alternating 12 h light-dark cycle. Food and water were available
Immobilization stress
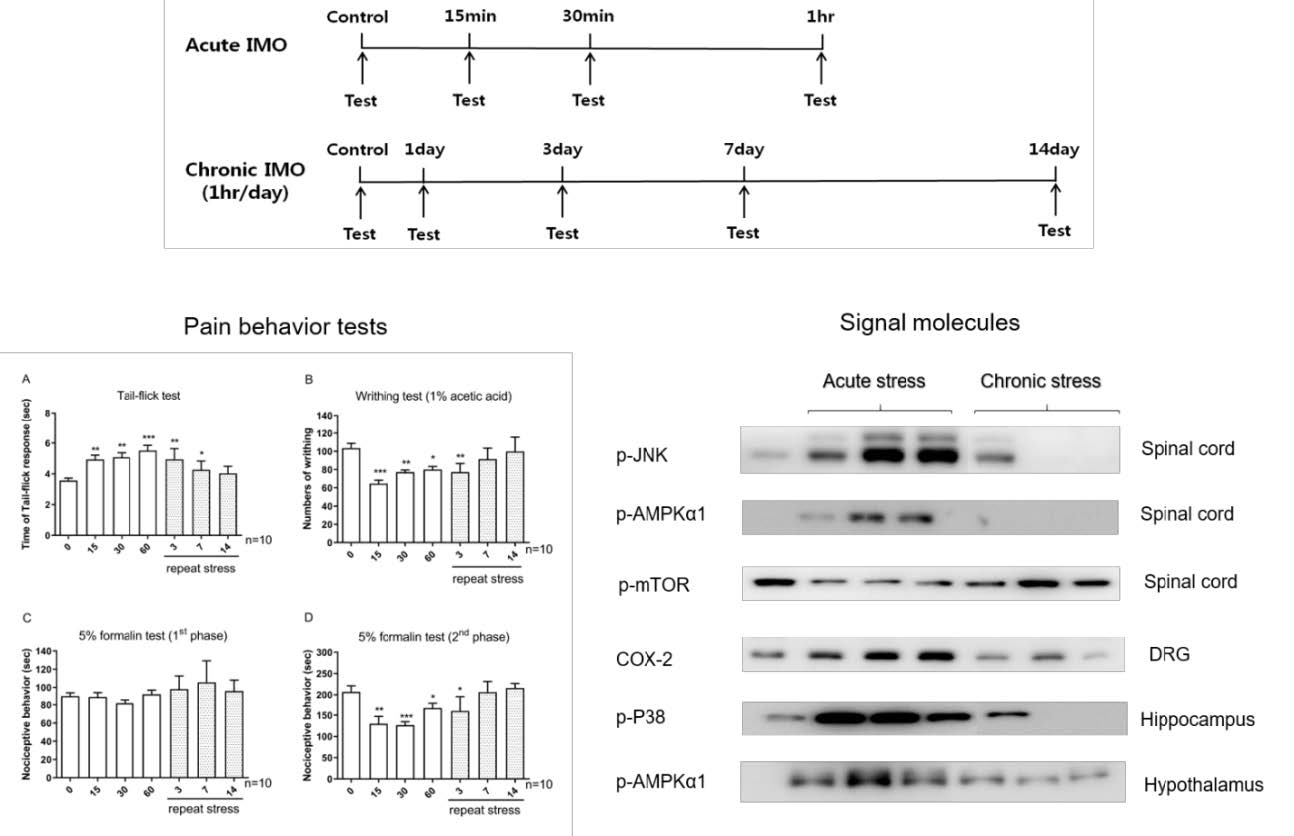
The mice were subjected to immobilization stress as described previously [20]. The mice were immobilized in 50 mL Corning tube and adjusting it with two iron nails on the outside, which crossed in the caudal part of the animal and restrained against back-and-forth movement. Adequate ventilation was provided using holes at the sides of the tubes. The acute IMO mice were stressed once in this manner for 15/30/60 min. The chronic IMO mice were repeatedly exposed to stress 1 h daily for 1/3/7/14 consecutive days. The design of experimental time is shown as Fig. 1.
Tail-flick test
Antinociception was determined by the tail-flick test [21]. To measure the latency of the tail-flick response, mice were gently held with one hand with the tail positioned in an apparatus (EM-DIE Instrument Co., Maidens, VA, U.S.A., Model TF6) for radiant heat stimulation. The tail-flick response was elicited by applying radiant heat to the dorsal surface of the tail. The intensity of heat stimulus in the tail-flick test was adjusted so that the animal flicked its tail within 3 to 5 s. In the absence of a withdrawal reflex, set the stimulus cutoff to 10 sec to avoid possible tissue damage.
Acetic acid-induced writhing
For the writhing test [22], 1% acetic acid was injection i.p. and then, the animals were immediately placed in an acrylic observation chamber (20 cm high, 20 cm diameter). The number of writhes was counted for 30 min after the injection of acetic acid. A writhe was defined as a contraction of the abdominal muscles accompanied by an extension of the forelimbs and elongation of the body.
Intraplantar formalin tests
For the formalin test [23], 10 μl of 5% formalin was injected subcutaneously under the plantar surface of the left hind paw. Following the injection of formalin, the animals were immediately placed in an acrylic observation chamber, and the time spent licking, shaking and biting the injected paw was measured with a stop-watch timer and considered as an indication of nociception. The first phase of the nociceptive response normally peaked 0 to 5min, and the second phase 20 to 40 min after formalin injection, representing the direct effect on nociceptors and inflammatory nociceptive responses, respectively [24].
Protein extraction and western blot
The lumbar section of the spinal cord of mice was dissected. Tissue was washed two times with cold Tris-buffered saline (20 mmol/L Trizma base and 137 mmol/L NaCl, pH 7.5). Immediately after washing, tissues were lysed with sodium dodecyl sulfate lysis buffer (62.5 mmol/L Trizma base, 2% w/v sodium dodecyl sulfate, 10% glycerol) containing 0.1 mmol/L Na3VO4, 3 mg/mL aprotinin, and 20 mmolL NaF. After brief sonication, the concentration of protein was determined with a detergent-compatible protein assay reagent (Bio-Rad Laboratories, Hercules, CA, USA) using bovine serum albumin as the standard. After adding bromophenol blue (0.1% w/v), the proteins were boiled, separated by electrophoresis in 6~10% polyacrylamide gels, and transferred onto a polyvinylidene difluoride membrane (Millipore, Bedford, MA, USA). The membranes were immunoblotted with antibodies p-JNK (Cell Signaling, 1:1000), p-AMPKα1 (Cell Signaling, 1:1000), p-mTOR (Cell Signaling, 1:1000), COX-2 (Abcam, 1:1000), p-P38 (Cell Signaling, 1:1000), and β-actin (Cell Signaling, 1:1000) in a blocking buffer for overnight. Membranes were then washed 4 times with Tris-buffered saline containing 20% Tween-20 (TBST; 10 mM Trizma base, pH8.0, 150 mM NaCl, and 20% Tween 20) for 20 min and then incubated with the anti-rabbit IgG-horseradish peroxidase conjugate (1:4000) in blocking buffer at room temperature for 1 h. After washing the membranes with TBST for 20 min (4 times), the antibody-antigen complexes were detected using the ECL system and exposed to Luminescent Image Analyzer (LAS-4000, Fuji Film Co., Tokyo, Japan) for the detection of light emission. p-JNK, p-AMPKα1, p-mTOR, COX-2, p-P38, and β-actin band densities were evaluated from the respective band densitometry. The Multi-Gauge Version 3.1 (Fuji Film Co., Tokyo, Japan) was used to analyze the intensity of expression. These values were expressed as the percentage of the control tested protein/β-actin for each sample.
Statistical analysis
Statistical analysis was assessed by one-way ANOVA with Bonferroni’s post-hoc test using GraphPad Prism Version 4.0 for Windows (GraphPad Software, San Diego, CA, USA). p-values less than 0.05 were considered to indicate statistical significance. All values were expressed as the mean±standard error.
RESULTS
Effect of acute and chronic immobilization stress on pain behavior in tail-flick, writhing, and formalin pain models
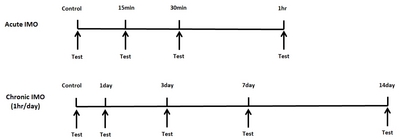
Mice were forced into acute IMO for 15, 30, or 60 min/day. In addition, in the chronic IMO group, mice were forced into 3, 7, 14 days (1 hr/day). Then the pain behavior was measured. In the tail-flick test, the tail-flick response was increased in 15 min and further increased by 60 min after acute IMO stress, as shown in Fig. 2A. However, the IMO-induced antinociceptive effect was gradually reduced after 3, 7, and 14 days. In the writhing test, the number of writhing responses was reduced 15, 30, and 60 min after acute IMO stress (Fig. 2B). In the chronic IMO stress group, the number of writhing responses recovered gradually to the normal level (Fig. 2B). In the formalin-induced pain model, either acute or chronic IMO stress did not alter pain behavior during the first phase as shown in Fig. 1C. However, the nociceptive behavior induced by formalin during the second phase was reduced by 15, 30, and 60 min after acute IMO stress (Fig. 2D). Additionally, the nociceptive behavior gradually increased toward the normal level (Fig. 2D).
Effect of acute and chronic immobilization stress on JNK, AMPKα1, mTOR protein phosphorylation in the spinal cord
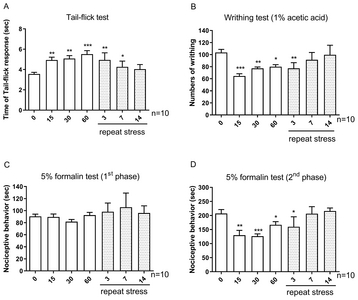
We examined the possible involvement of p-JNK, p-AMPKα1 and p-mTOR proteins in IMO stress-induced antinociception in the spinal cord. As shown in Fig. 2A, and 2B, acute IMO stress gradually increased p-JNK, p-AMPKα1 protein expression in the spinal cord 15, 30, and 60 min after the stress stimulation. However, the increased phosphorylation of JNK and AMPKα1 proteins induced by acute IMO stress were gradually decreased on 3, 7, and 14 days after IMO stress stimulation (Fig. 3A and 3B). In contrast to the expression of p-AMPKα1, p-mTOR protein expression was downregulated after 15, 30, and 60 min after acute IMO stress stimulation. However, downregulated p-mTOR protein expression induced by acute IMO stress was gradually increased on 3, 7, and 14 days after IMO stress stimulation in the spinal cord (Fig. 3C).
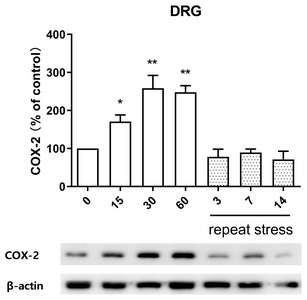
Effect of acute and chronic immobilization stress on COX-2 protein expression in DRG
In order to observe the change of COX-2 protein expression in IMO stress-induced antinociception in DRG, COX-2 protein was extracted from anatomical DRG after IMO stress. As shown in Fig. 4, COX-2 protein expression was upregulated after 15, 30, and 60 min after acute IMO stress stimulation. However, upregulated COX-2 protein expression induced by acute IMO stress was gradually decreased on 3, 7, and 14 days after IMO stress stimulation in DRG (Fig. 4).
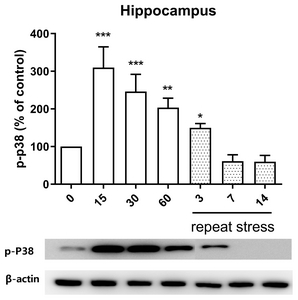
Effect of acute and chronic immobilization stress on P38 protein phosphorylation expression in the hippocampus
We examined the possible role of P38 protein phosphorylation expression in hippocampus and found that the acute IMO stress stimulation for 15, 30 or 60 min enhanced the phosphorylation expression of P38 protein in the hippocampus, but upregulated expression of phosphorylated P38 protein induced by acute IMO stress was gradually reduced on 3, 7, and 14 days by chronic stress stimulation (Fig. 5).
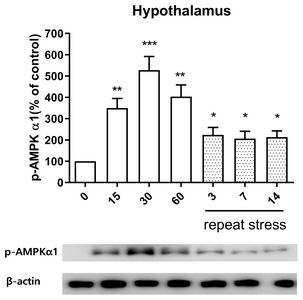
Effect of acute and chronic immobilization stress on AMPKα1 protein phosphorylation expression in the hypothalamus
In the hypothalamus, the pattern of p-AMPKα1 protein expression was similar to its expression in the spinal cord. The phosphorylation expression of AMPKα1 protein was increased at 15, 30, and 60 min after acute IMO stress stimulation in the hypothalamus (Fig. 6). However, the increased expression of p-AMPKα1 proteins induced by acute IMO stress was gradually decreased on 3, 7, and 14 days by chronic stress stimulation (Fig. 6).
DISCUSSION
In the present study, the effects of acute and chronic IMO stress on the pain behavior in various pain models were examined and we found that acute IMO stress exerts antinociceptive effects in tail-flick, writhing, and formalin pain models. These results are similar to the findings reported previously. For example, Gamaro et al. [25] have previously reported that acute immobilization stress produced an antinociceptive effect in the rat in the tail-flick test. In addition, Heidari-Oranjaghi et al. [3] found that acute restraint stress also produced analgesia in the formalin test. We further examined if chronic IMO stress alters the antinociceptive effects induced by acute IMO stress. We found in the present study that chronic IMO stress causes antinociceptive tolerance as revealed in tail-flick, writhing, and formalin pain tests. Our result is in part in line with the study observed by Gamaro et al. group [25]. They found that female mice showed similar performance as the control group after 1 h per day for 40 days of restraint stress, while chronically stressed male rats decreased tail-flick latency. The results of the present study clearly suggest that chronic IMO stress causes antinociceptive tolerance. As with a series of studies that have reported immobilization SIA being observed only in the second phase of formalin response [5, 20, 26], in this study, we also find that both single and repeated immobilization stress attenuated only the second phase of formalin response, but not the first phase. It has been known that the intraplantar injection produces a biphasic reaction [27]. The pain behavior observed during first phase is mostly due to the direct stimulation of nociceptors, whereas the pain behaviors observed during the second phase involves both inflammatory mechanisms and central sensitization within the dorsal horn [28], suggesting that pain behaviors observed during the first and second phases are differentially regulated.
Previously, we have indicated that long-term repeated immobilization stress (more than 7 days) shows the differential tolerance on antinociceptive effect induced by a single immobilization stress by using several pain models, and we suggested that the tolerance of stress-induced analgesia induced by repeated stresses may be due to β-endorphin and 5-hydroxytryptamine [5, 20]. In addition, we also suggested that repeatedly 7 times cold water swimming stress (4°C, 3 min) develops a tolerance on the analgesia induced by single cold water swimming stress, which might be induced by the delta-opioid receptor [29]. Moreover, Fazli-Tabaei et al. [30] found that 3 days of exposure to swim stress (20°C, 3 min) induces tolerance to swim stress-induced antinociception in the second phase of the formalin test and the D2 dopamine receptor may be associated with the different effect of single and repeated swim stress. Furthermore, Pavlovic and Bodnar [31] reported that 14 days continuous cold-water swims (2°C, 3 min) and 14 days intermittent cold-water swims (ICWS: 2°C, 18 10-s swims, 18 10-s recoveries) display tolerance to the antinociception induced by cold water swim. Also, they suggested that the development of tolerance of antinociception may be an environmentally induced representation of delta2- and mu-opioid receptors. Although the exact mechanisms of antinociception induced by acute or chronic IMO stress are currently not clear, these lines of evidence have demonstrated that might be due to changes in multiple descending pain inhibitory systems.
Several signal molecules such as JNK, AMPKα1, and mTOR proteins are known to play important roles in the regulation of pain or antinociception [32–34]. However, the relationship of the expression of JNK, AMPKα1 and mTOR proteins and acute stress-induced antinociception in the spinal cord has not been reported. In the present study, we found that acute IMO stress causes elevations of phosphorylated JNK and AMPKα1 molecules but chronic IMO stress reduces acute IMO stress-induced enhanced phosphorylated JNK, AMPKα1 protein levels, suggesting that JNK and AMPKα1 molecules located in the spinal cord play important roles in the regulation both of the antinociception by acute IMO stress and of the development of the antinociceptive tolerance by chronic IMO stress. In addition, Kim et al. [35] demonstrated that levels of AMPK gene expression in the rat hypothalamus determine the sensitivity to electroacupuncture (EA) analgesia. Thus, our findings also suggest that the phosphorylated AMPKα1 expression in the hypothalamus appears to be responsible for an increase of the antinociceptive effect by acute IMO stress. Moreover, the phosphorylation of AMPKα1 might be also responsible for the production of tolerance on acute IMO stress-induced antinociception after chronic IMO stress. On the other hand, we found that mTOR display beneficial effects in the regulation of the antinociception by acute IMO stress, which is also characterized by the development of antinociceptive tolerance by chronic IMO stress. Our findings are consistent with previous studies that inhibitors of mTOR are identified as a potential target of chronic inflammatory pain [36] and neuropathic pain [17, 37].
Although several previous studies reported that COX-2 plays a major role in the inflammatory pain and neuropathic pain [38–40], whether COX2 is associated with antinociceptive effects by IMO stress has not been known. Our current finding that COX-2 located in DRG is upregulated by acute IMO stress suggests that COX-2 is also associated with the production of antinociception induced by acute IMO stress. Furthermore, the reduction of acute-IMO stress-induced upregulation of COX-2 protein expression in DRG by chronic IMO also suggests that COX-2 protein in DRG might play an important role in the development of the antinociceptive tolerance by chronic IMO stress.
Recently, several studies have demonstrated that the expression and activity of P38 in several brain regions that are components of the descending pain modulatory system [41, 42]. Expression of P38 located in the hippocampus is changed by acute stress, such as glucocorticoid stress, electroacupuncture, and hyperosmotic stress. Grassmé et al. [43] have reported that glucocorticoid stress increases the phosphorylation of P38 protein in the hippocampus. Xu et al. [44] have demonstrated that electroacupuncture (EA) increased the phosphorylation of P38 protein in the hippocampus. Hyperosmotic stress activated the phosphorylation of p38 MAPK in the hippocampus [45]. These findings suggest that P38 plays an important role in acute stress-induced antinociception. In support of these findings, in the present study, we found that acute IMO stress also causes elevations of phosphorylated P38 molecule, which suggests that P38 protein appears to be associated with the production of antinociception induced by acute IMO stress. Moreover, we found that chronic IMO stress reduces IMO-induced upregulation of phosphorylated P38 protein level, suggesting that P38 plays an important role in the regulation of the development of the antinociceptive tolerance by chronic IMO stress.
Taken together, our results suggest that the chronic IMO stress causes the development of tolerance to the antinociception induced by acute IMO stress. Moreover, the COX-2 in DRG, p-JNK, p-AMPKα1, and p-mTOR in the spinal cord, p-P38 in the hippocampus, and p-AMPKα1 in the hypothalamus may play important roles in the regulation of antinociception induced by acute IMO stress and the antinociceptive tolerance development induced by chronic IMO stress.
ACKNOWLEDGEMENTS
This research was supported by Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Science, ICT & Future Planning (2017M3C7A1020062).
Figures
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
References
- Amit Z, Galina ZH (1986) Stress-induced analgesia: adaptive pain suppression. Physiol Rev 66:1091-1120.


- Butler RK, Finn DP (2009) Stress-induced analgesia. Prog Neurobio 88:184-202.
- Heidari-Oranjaghi N, Azhdari-Zarmehri H, Erami E, Haghparast A (2012) Antagonism of orexin-1 receptors attenuates swim- and restraint stress-induced antinociceptive behaviors in formalin test. Pharmacol Biochem Behav 103:299-307.
- Porro CA, Carli G (1988) Immobilization and restraint effects on pain reactions in animals. Pain 32:289-307.
- Seo YJ, Kwon MS, Shim EJ, Park SH, Choi OS, Suh HW (2006) Changes in pain behavior induced by formalin, substance P, glutamate and pro-inflammatory cytokines in immobilizationinduced stress mouse model. Brain Res Bull 71:279-286.
- Pezalla PD, Dicig M (1984) Stress-induced analgesia in frogs:evidence for the involvement of an opioid system. Brain Res 296:356-360.
- Yang X, Guo Z, Lu J, Zhao B, Fei Y, Li J, Jiang H, Sun L, Wang Y, Sun Y, Bao T (2017) The role of MAPK and dopaminergic synapse signaling pathways in antidepressant effect of electroacupuncture pretreatment in chronic restraint stress rats. Evid Based Complement Alternat Med 2017:2357653.

- Chen J, Zhao M, Rao R, Inoue H, Hao CM (2005) C/EBP{beta} and its binding element are required for NF{kappa} B-induced COX2 expression following hypertonic stress. J Biol Chem 280:16354-16359.
- Madrigal JL, García-Bueno B, Caso JR, Pérez-Nievas BG, Leza JC (2006) Stress-induced oxidative changes in brain. CNS Neurol Disord Drug Targets 5:561-568.
- Sakai S, Hagihara N, Kuse M, Kimura K, Okuda K (2018) Heat stress affects prostaglandin synthesis in bovine endometrial cells. J Reprod Dev 64:311-317.
- Bayir H, Kagan VE, Borisenko GG, Tyurina YY, Janesko KL, Vagni VA, Billiar TR, Williams DL, Kochanek PM (2005) Enhanced oxidative stress in iNOS-deficient mice after traumatic brain injury: support for a neuroprotective role of iNOS. J Cereb Blood Flow Metab 25:673-684.
- Lerouet D, Beray-Berthat V, Palmier B, Plotkine M, Margaill I (2002) Changes in oxidative stress, iNOS activity and neutrophil infiltration in severe transient focal cerebral ischemia in rats. Brain Res 958:166-175.
- Reuter S, Gupta SC, Chaturvedi MM, Aggarwal BB (2010) Oxidative stress, inflammation, and cancer: how are they linked? Free Radic Biol Med 49:1603-1616.
- Asiedu MN, Dussor G, Price TJ (2016) Targeting AMPK for the alleviation of pathological pain. Exp Suppl 107:257-285.
- Lin JP, Chen CQ, Huang LE, Li NN, Yang Y, Zhu SM, Yao YX (2018) Dexmedetomidine attenuates neuropathic pain by inhibiting P2X7R expression and ERK phosphorylation in rats. Exp Neurobiol 27:267-276.
- Cui J, He W, Yi B, Zhao H, Lu K, Ruan H, Ma D (2014) mTOR pathway is involved in ADP-evoked astrocyte activation and ATP release in the spinal dorsal horn in a rat neuropathic pain model. Neuroscience 275:395-403.
- Lisi L, Aceto P, Navarra P, Dello Russo C (2015) mTOR kinase:a possible pharmacological target in the management of chronic pain. Biomed Res Int 2015:394257.
- Lutz BM, Nia S, Xiong M, Tao YX, Bekker A (2015) mTOR, a new potential target for chronic pain and opioid-induced tolerance and hyperalgesia. Mol Pain 11:32.
- Zhang W, Sun XF, Bo JH, Zhang J, Liu XJ, Wu LP, Ma ZL, Gu XP (2013) Activation of mTOR in the spinal cord is required for pain hypersensitivity induced by chronic constriction injury in mice. Pharmacol Biochem Behav 111:64-70.
- Seo YJ, Kwon MS, Choi SM, Lee JK, Park SH, Jung JS, Sim YB, Suh HW (2011) Differential cross-tolerance development between single and repeated immobilization stress on the antinociceptive effect induced by β-endorphin, 5-hydroxytryptamine, morphine, and WIN55,212-2 in the inflammatory mouse pain mode. Arch Pharm Res 34:269-280.
- Horan P, Taylor J, Yamamura HI, Porreca F (1992) Extremely long-lasting antagonistic actions of nor-binaltorphimine (nor-BNI) in the mouse tail-flick test. J Pharmacol Exp Ther 260:1237-1243.
- Koster R (1959) Acetic acid for analgesic screening. Fed Proc 18:412.
- Hunskaar S, Fasmer OB, Hole K (1985) Formalin test in mice, a useful technique for evaluating mild analgesics. J Neurosci Methods 14:69-76.
- Hunskaar S, Hole K (1987) The formalin test in mice: dissociation between inflammatory and non-inflammatory pain. Pain 30:103-114.
- Gamaro GD, Xavier MH, Denardin JD, Pilger JA, Ely DR, Ferreira MB, Dalmaz C (1998) The effects of acute and repeated restraint stress on the nociceptive response in rats. PhysiolBehav 63:693-697.
- Vaccarino AL, Marek P, Liebeskind JC (1992) Stress-induced analgesia prevents the development of the tonic, late phase of pain produced by subcutaneous formalin. Brain Res 572:250252.
- Barrot M (2012) Tests and models of nociception and pain in rodents. Neuroscience 211:39-50.
- Tjølsen A, Berge OG, Hunskaar S, Rosland JH, Hole K (1992) The formalin test: an evaluation of the method. Pain 51:5-17.
- Kim KW, Choi SS, Woo RS, Suh HW (2003) Development of antinociceptive tolerance and changes of opioid receptor ligand binding in central nervous system of the mouse forced to single and repeated swimming in the cold water. Brain Res Bull 61:93-97.
- Fazli-Tabaei S, Yahyavi SH, Nouri M, Zartab H, Javid G, Loghavi S, Zarrindast MR (2006) Dopamine receptor mechanism(s) and antinociception and tolerance induced by swim stress in formalin test. Behav Pharmacol 17:341-347.
- Pavlovic Z, Bodnar RJ (1993) Antinociceptive and hypothermic crosstolerance between continuous and intermittent cold-water swims in rats. Physiol Behav 54:1081-1084.
- Gao YJ, Ji RR (2008) Activation of JNK pathway in persistent pain. Neurosci Lett 437:180-183.
- Ge A, Wang S, Miao B, Yan M (2018) Effects of metformin on the expression of AMPK and STAT3 in the spinal dorsal horn of rats with neuropathic pain. Mol Med Rep 17:5229-5237.
- Xu JT, Zhao JY, Zhao X, Ligons D, Tiwari V, Atianjoh FE, Lee CY, Liang L, Zang W, Njoku D, Raja SN, Yaster M, Tao YX (2014) Opioid receptor-triggered spinal mTORC1 activation contributes to morphine tolerance and hyperalgesia. J Clin Invest 124:592-603.
- Kim SK, Sun B, Yoon H, Lee JH, Lee G, Sohn SH, Kim H, Quan FS, Shim I, Ha J, Min BI, Bae H (2014) Expression levels of the hypothalamic AMPK gene determines the responsiveness of the rats to electroacupuncture-induced analgesia. BMC Complement Altern Med 14:211.
- Liang L, Tao B, Fan L, Yaster M, Zhang Y, Tao YX (2013) mTOR and its downstream pathway are activated in the dorsal root ganglion and spinal cord after peripheral inflammation, but not after nerve injury. Brain Res 1513:17-25.
- Kwon M, Han J, Kim UJ, Cha M, Um SW, Bai SJ, Hong SK, Lee BH (2017) Inhibition of mammalian target of rapamycin (mTOR) signaling in the insular cortex alleviates neuropathic pain after peripheral nerve injury. Front Mol Neurosci 10:79.
- Ma W, Chabot JG, Vercauteren F, Quirion R (2010) Injured nerve-derived COX2/PGE2 contributes to the maintenance of neuropathic pain in aged rats. Neurobiol Aging 31:12271237.
- Ma W, Quirion R (2008) Does COX2-dependent PGE2 play a role in neuropathic pain? Neurosci Lett 437:165-169.
- Vardeh D, Wang D, Costigan M, Lazarus M, Saper CB, Woolf CJ, Fitzgerald GA, Samad TA (2009) COX2 in CNS neural cells mediates mechanical inflammatory pain hypersensitivity in mice. J Clin Invest 119:287-294.
- Imbe H, Senba E, Kimura A, Donishi T, Yokoi I, Kaneoke Y (2011) Activation of mitogen-activated protein kinase in descending pain modulatory system. J Signal Transduct 2011:468061.
- Mizushima T, Obata K, Yamanaka H, Dai Y, Fukuoka T, Tokunaga A, Mashimo T, Noguchi K (2005) Activation of p38 MAPK in primary afferent neurons by noxious stimulation and its involvement in the development of thermal hyperalgesia. Pain 113:51-60.
- Grassmé H, Jernigan PL, Hoehn RS, Wilker B, Soddemann M, Edwards MJ, Müller CP, Kornhuber J, Gulbins E (2015) Inhibition of acid sphingomyelinase by antidepressants counteracts stress-induced activation of P38-kinase in major depression. Neurosignals 23:84-92.
- Xu J, She Y, Su N, Zhang R, Lao L, Xu S (2015) Effects of electroacupuncture on chronic unpredictable mild stress rats depression-like behavior and expression of p-ERK/ERK and p-P38/P38. Evid Based Complement Alternat Med 2015:650729.
- Niswander JM, Dokas LA (2007) Hyperosmotic stressinduced caspase-3 activation is mediated by p38 MAPK in the hippocampus. Brain Res 1186:1-11.