Articles

Article Tools
 View Full Text View Full Text |
 Abstract Abstract |
 Article as PDF Article as PDF |
 Print this Article Print this Article |
 Pubmed Pubmed |
 PMC PMC |
 PubReader PubReader |
 Export to Citation Export to Citation |
 Email Alerts Email Alerts |
 Open Access Open Access |
Share this article on :



Stats or Metrics
Article
Original Article
Exp Neurobiol 2019; 28(4): 504-515
Published online August 31, 2019
https://doi.org/10.5607/en.2019.28.4.504
© The Korean Society for Brain and Neural Sciences
HDAC Inhibition by Valproic Acid Induces Neuroprotection and Improvement of PD-like Behaviors in LRRK2 R1441G Transgenic Mice
Taewoo Kim†, Seohoe Song†, Yeongwon Park, Sinil Kang and Hyemyung Seo*
Correspondence to: *To whom correspondence should be addressed.
TEL: 82-31-400-5511, FAX: 82-31-419-1760
e-mail: hseo@hanyang.ac.kr
†These authors contributed equally to this work.
This is an Open Access article distributed under the terms of the Creative Commons Attribution Non-Commercial License(http://creativecommons.org/licenses/by-nc/4.0) which permits unrestricted non-commercial use, distribution, andreproduction in any medium, provided the original work is properly cited.
Abstract
Parkinson’s disease (PD) is one of the late-onset neurodegenerative movement disorder. Major pathological markers of PD include progressive loss of dopaminergic neurons, Lewy body formation, genetic mutations, and environmental factors. Epigenetic regulation of specific gene expression via impaired histone acetylation is associated with neuronal dysfunction in various neurodegenerative diseases. In this study, we hypothesized that histone deacetylase (HDAC) inhibitor, valproic acid (VPA), can improve motor function by enhancing cell survival in PD genetic model mice with LRRK2 R1441G mutation. To address this question, we administered VPA in LRRK2 R1441G transgenic mice to determine whether VPA affects 1) histone acetylation and HDAC expression, 2) dopaminergic neuron survival, 3) inflammatory responses, 4) motor or non-motor symptoms. As results, VPA administration increased histone acetylation level and the number of tyrosine hydroxylase (TH) positive neurons in substantia nigra of LRRK2 R1441G mice. VPA reduced iba-1 positive activated microglia and the mRNA levels of pro-inflammatory marker genes in LRRK2 R1441G mice. In addition, VPA induced the improvement of PD-like motor and non-motor behavior in LRRK2 R1441G mice. These data suggest that the inhibition of HDAC can be further studied as potential future therapeutics for PD.
Graphical Abstract
Keywords: Parkinson&rsquo,s disease (PD), Histone deacetylase (HDAC), Valproic acid (VPA), Neuroprotection
INTRODUCTION
Parkinson’s disease (PD) patients show motor dysfunction including rigidity, tremor, postural instability, and bradykinesia and non-motor dysfunction in emotional control such as depression and anxiety. The pathological hallmarks of PD include loss of dopaminergic (DA) neurons in the substantia nigra and the presence of Lewy bodies composed of highly stable α-synuclein (α-syn) in PD patients’ brains [1, 2]. Familial PD is associated with several specific gene mutations including leucine-rich repeat kinase (LRRK2), α-syn, Parkin, PTEN-induced putative kinase 1 (PINK1), DJ-1 and more [3–7].
Previous studies have revealed pathophysiological role of LRRK2 in PD and a strong correlation between LRRK2 and inflammatory responses. Several evidences suggested that LRRK2 could act as a modulator of inflammatory response through toll like receptor (TLR) family signal pathway [8, 9]. LRRK2 R1441G mutation increased tumor necrosis factor-α (TNF-α) release and decreased inhibitory cytokine interleukin-10 (IL-10) secretion in LPS-stimulated microglial cells [10]. Moreover, inhibition of LRRK2 or knockdown of LRRK2 decreased the secretion of TNF-α in rat primary microglia [11]. These evidences suggest that LRRK2 has important role in pathological process of PD through inflammatory mechanism and LRRK2 mutation may induce abnormal inflammatory responses in PD.
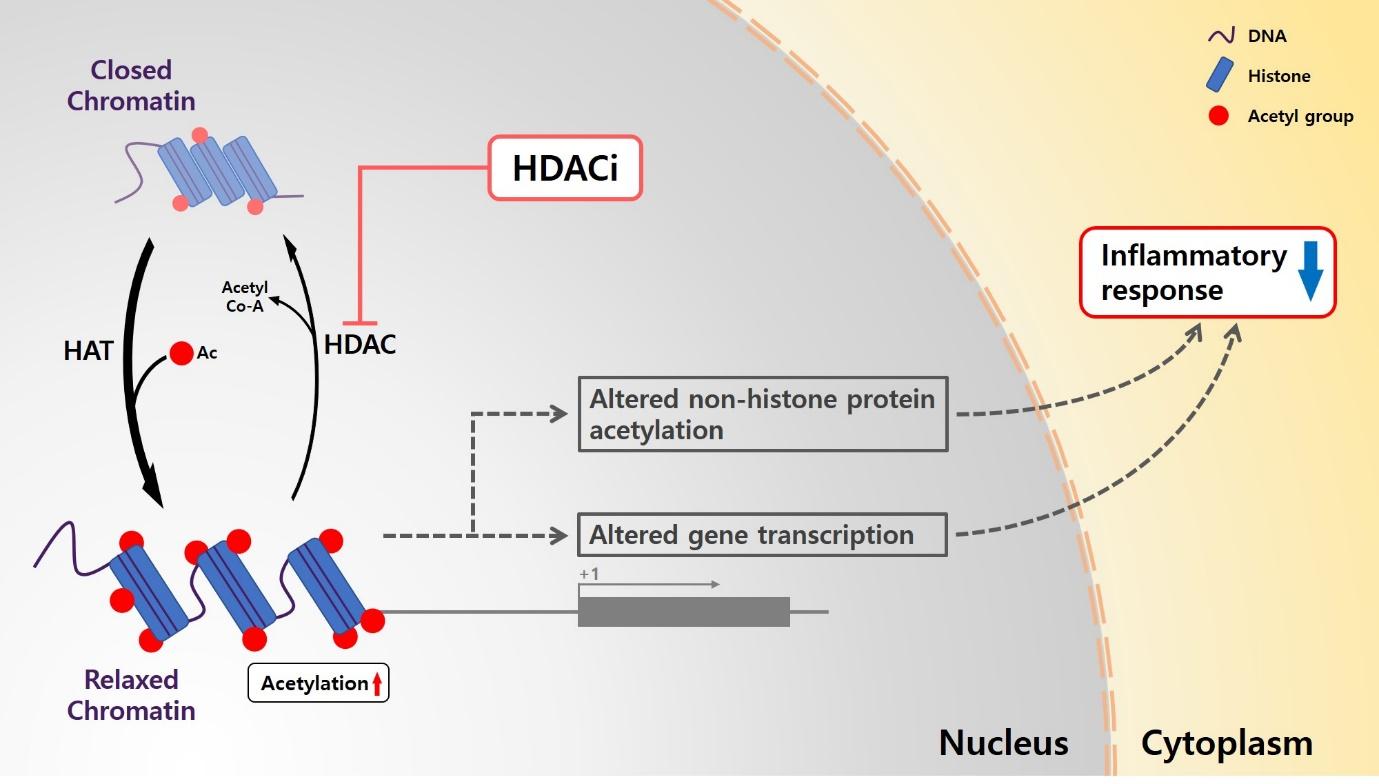
Histone acetylation is a dynamic process which is catalyzed by two enzymes histone acetyltransferase (HAT) and histone deacetylase (HDAC) [12, 13]. The acetylation or deacetylation of histone N-terminal tails allows for control of accessibility to DNA by altering the interaction between histones and DNA sequences in the chromatin [14]. In general, this hyperacetylation of histone residue is strongly related to relaxing the chromatin structure, thus leading it to be more accessible to transcriptional activation [15]. Acetylated histone or non-histone protein exert their neuroprotective effects by reducing inflammatory response and inhibiting neuronal cell death [16–18]. Regulation of acetylation level of histone could improve neuronal functions in many neurological disorders such as cerebral ischemia, spinal cord injury, and traumatic brain injury [19–21].
Several previous studies demonstrated that various HDAC inhibitors, including valproic acid (VPA), class I/IIa HDAC inhibitor, caused hyperacetylation of histone or non-histone proteins and altered the physiological condition of dopaminergic neurons. VPA, SB, and TSA dose-dependently increased the amount of dopamine (DA) uptake in rat primary mesencephalic neuron-glia cultures [22, 23]. VPA decreased the number of microglial cells and the protein level of TNF-α, and oxidative stress level in a time- and dose-dependent manner in rat primary microglia-enriched culture [24]. VPA increased DA uptake and the number of tyro-sine hydroxylase (TH)-positive neurons in rat primary mesencephalic neuron-glia culture [24]. SB, VPA, and SAHA, all induced hyperacetylation of histone 3 at Lys 9 and resulted in significant increase in cell viability and increased the number of TH-positive cells under MPP+ treatment. Moreover, the administration of VPA in MPTP-treated mice also resulted in increased levels of acetylated histone 3 at Lys 9 and significantly increased levels of striatal dopamine [25]. In this study, we hypothesized that VPA reveals the neuroprotective effect through inflammatory control, which improves PD-like behaviors in genetic PD model containing LRRK2 R1441G mutation.
MATERIALS AND METHODS
Experimental animals and VPA administration
LRRK2 R1441G transgenic mice (FVB/N-Tg 135Cjli/J) were purchased from Jackson Laboratory (Bar Harbor, ME) [26–28]. The mice were maintained on a 12-h light/dark cycle and fed
Rota-rod test
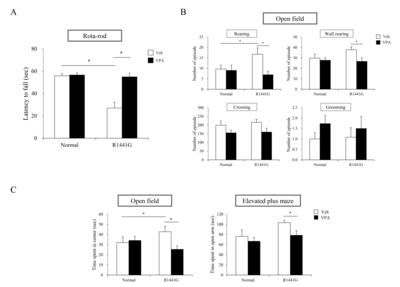
To determine motor ability and balancing, all mice were tested in the rota-rod behavioral assessments before and after VPA injection as previously described [30]. In the test, mice were placed on the rota-rod (Panlab, Barcelona, Spain) for 60 seconds at the speed of 5 rpm for 4 trials. Latency to fall (sec) was recorded for each mouse at each trial.
Open filed test
To check motor coordination, exploratory behavior, and anxiety-like behavior, experimental mice were assessed in the open field test [31]. Mice were placed in the center of a transparent acrylic box (27.5×27.5 cm) and allowed to move freely. The bottom of acrylic box was divided into 25 squares (5.5×5.5 cm) with black lines. Mice were submitted to 3 consecutive trials and each trial was for 4 min. During the test, movements (line crossing, rearing, wall rearing, grooming) and time spent in center 5 squares were counted and recorded for each mouse in each trial. Line crossing is the number of movements in which both hind legs crossed the black line completely. The number of rearing was counted when the mouse stood up by its hind legs. The number of wall rearing was movement in which the mouse stood up and leaned on the wall and placed their forelegs on the wall. The number of grooming was counted when the mouse was keeping itself clean and grooming its facial or body hair.
Elevated plus maze test
To check anxiety-like behavior, experimental mice were tested on the elevated plus maze test as previous described [32]. As short description, the elevated plus maze consisted of two 30×5 cm open arms, and two 30×5×15.25 cm closed arms with open roof, elevated at a height of 40 cm. The four arms extended from a common center area platform (5×5 cm). Each mouse was placed on the center area, facing the same open arm and allowed to explore freely for 2 min, one day before behavioral assessments. For elevated plus maze test, mice were placed on the same center area in the same pattern with free exploration session and time spent and number of entries into each area were recorded for 5 min.
RNA preparation
RNA samples were extracted from freshly dissected brain tissue using TRI Reagent TM (Sigma, St. Louis, MO) according to the manufacturer’s instruction. After adding 500 μl of TRI Reagent, the cell lysate was homogenized by hand-held homogenizer and incubated for 5 minutes. Chloroform, 100 μl (Sigma, St. Louis, MO) was added into each sample and mixed with vigorous shaking for 15 seconds. After incubation for 10 minutes, mixture was centrifuged at 12,000×g for 15 minutes at 4°C and the aqueous phase was transferred to a fresh tube. Then, 500 μl of 2-propanol per ml of TRI reagent was added and mixed, and then the samples were centrifuged at 12,000×g for 15 min at 4°C. Supernatants were removed, and the RNA pellet was washed by adding 500 μl of 75% ethanol. The samples were vortexed and then centrifuged at 7,500×g for 5 min at 4°C. The supernatant was removed, and the RNA pellet was dried for 20 min. After drying, the RNA pellet was dissolved in DEPC-treated water (Ambion, Austin, TX) and stored at −20°C until use.
Reverse transcription polymerase chain reaction (RT-PCR)
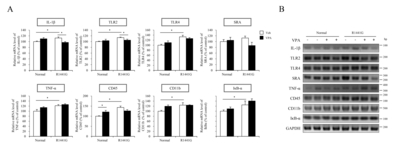
RNA samples were transcribed into cDNA using Premium express 1st strand cDNA synthesis system (Legene, San Diego, CA) according to the manufacturer’s instruction. In brief, a total 500 ng of RNA sample were incubated at 65°C with Oligo (dT). RNA/primer mix were incubated at 37°C for 50 min after adding reaction mixture. For inactivation of reverse transcriptase, cDNA samples were heated at 85°C for 5 min and stored at −20°C until use. The primers which were used to amplify the target genes from the cDNA are shown in Table 1. PCR was performed with G-Taq DNA polymerase (Labopass, Seoul, Korea) using the following conditions: denaturation step at 94°C for 2 min, followed by 30 cycles of 30 sec at 94°C, 1 min at 58°C, and 30 sec at 72°C, and termination step at 72°C for 7 min. PCR products were visualized in 2.5% agarose gel electrophoresis and detected by print-graph image machine (ATTO, Tokyo, Japan). Each visualized band intensity was quantified by Image J program (version 1.46r; NIH, Bethesda, MD) and normalized with the GAPDH expression level.
Protein preparation and western blot
Protein samples were prepared from freshly dissected brain and used for semi-quantitative western blot analysis as previously described [33]. Primary antibodies were used in following conditions: anti-histone H3 (1:2,000; Abcam, Cambridge, MA), anti-acetyl histone H3 (1:2,000; Abcam, Cambridge, MA), anti-β-actin (1:10,000; Abcam, Cambridge, MA), anti-HDAC1 (1:2,000; Cell Signaling Technology, Danvers, MA), anti-HDAC2 (1:2,000; Cell Signaling Technology, Danvers, MA), anti-HDAC3 (1:2,000; Cell Signaling Technology, Danvers, MA), anti-iba1 (1:2,000; Wako, Osaka, Japan). Properly matched secondary antibodies were used before ECL reaction: anti-mouse IgG (1:2,000; Vector Laboratories, Burlingame, CA), anti-rabbit IgG (1:10,000; Vector Laboratories, Burlingame, CA).
Immunohistochemistry
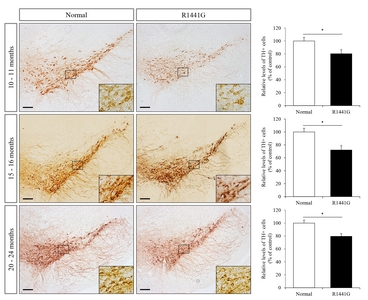
Immunohistochemistry was performed on brain sections (30 μm), which prepared using microtome (Thermo Fisher Scientific, Rockford, IL) to detect dopaminergic neurons or activated microglial cells in the brain regions of LRRK2 R1441G mice (Fig. 1). The representative brain sections were selected with reference to a coronal atlas of the mouse brain [34, 35]. We used rabbit antiTH (1:400; Pelfreez, Rogers, AR) or rabbit anti-ionized calcium-binding adapter molecule 1 (iba-1) (1:300; Wako, Osaka, Japan) primary antibodies. In brief, brain sections were blocked with 5% normal goat serum and 0.5% triton X-100 in 10mM PBS for 1 hour and incubated with each primary antibody for overnight. Sections were incubated with horseradish peroxidase (HRP)-linked secondary antibody and the signal was visualized using the DAB peroxidase substrate Kit (Vector Laboratories, Burlingame, CA). For quantification, images were taken under bright field illumination using Nikon Eclipse 80i microscope (Nikon, Tokyo, Japan). From three sections for each experimental mouse, the number of immune-reactive cells was blindly counted by 2 different investigators modified from previously reported cell counting methods [34].
Statistical analysis
All statistical analyses of behavioral tests and biochemical experiments were carried out using SPSS (version 21; IBM Corp, Armonk, NY). The data analyses were performed using the student’s t-test and ANOVA analysis. A p-value less than 0.05 was considered statistically significant.
RESULTS
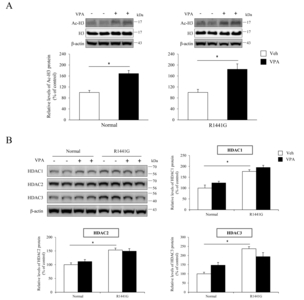
VPA administration increased histone acetylation level without change of HDAC expression in LRRK2 R1441G mice
We administered VPA, HDAC inhibitor, into LRRK2 R1441G transgenic mice to determine whether VPA induces neuroprotection and behavioral improvement in
VPA administration restored the number of dopaminergic neurons in the substantia nigra of LRRK2 R1441G mice
To determine the effect of VPA on the survival of dopaminergic neurons in the substantia nigra, we performed immunohistochemistry (IHC) staining with anti-tyrosine hydroxylase (TH) antibody (Fig. 3A). LRRK2 R1441G transgenic mice showed a significant decrease in the number of TH-positive dopaminergic neurons in the substantia nigra compared to littermate normal control mice. Interestingly, administration of VPA significantly increased the number of TH-positive dopaminergic neurons in LRRK2 R1441G transgenic mice. These data suggest that HDAC inhibition by VPA could exert neuroprotective effect on dopaminergic neurons in LRRK2 R1441G PD model mice.
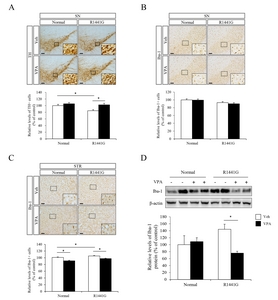
VPA administration attenuated the neuroinflammatory responses in LRRK2 R1441G mice
In the previous study, we identified that administration of VPA reduced the mRNA expression of proinflammatory genes including NF-κB, IL-1β and TLR4 in blood plasma or hippocampus of Tg6799 AD model mice at 10 months of age [37]. To address the effect of transcriptional regulation by VPA on neuroinflammatory response under LRRK2 R1441G mutation, we determined the population of activated microglia cells in substantia nigra (SN) or striatum (STR) by IHC staining with ionized calcium binding adaptor molecule-1 (iba-1) antibody (Fig. 3B and 3C). We found that VPA did not alter the number of the activated microglia in substantia nigra of both LRRK2 R1441G and littermate normal control mice. However, in the striatum, which is the projection area of nigrostriatal dopaminergic pathway, the number of activated microglia was significantly increased in LRRK2 R1441G mice compared to littermate normal control mice and VPA significantly decreased the number of the activated microglia in the striatum of LRRK2 R1441G and littermate normal control mice (Fig. 3C). We also determined the expression level of iba-1 protein in the cortical projection area of nigral DA neurons and found that VPA significantly decreased the iba-1 expression in LRRK2 R1441G mutant mice (Fig. 3D).
We next determined mRNA expression levels of several inflammatory markers by semi-quantitative RT-PCR in the midbrain regions of LRRK2 R1441G and normal control mice (Fig. 4). Remarkably, LRRK2 R1441G mice showed a significantly increased expression of inflammatory response related genes including IL-1β, TLR2, TLR4, CD45, CD11b, TNF-α, IκB-α, compared to littermate normal control mice (Fig. 4). Moreover, VPA decreased several inflammatory markers including IL-1β and TLR2 in LRRK2 R1441G mice compared to vehicle group. These data suggest that HDAC inhibition by VPA attenuated neuroinflammatory responses that were elevated in LRRK2 R1441G PD model mice.
VPA administration improved behavioral deficits in LRRK2 R1441G mutant mice
To determine VPA effects on the motor or non-motor PD-like behaviors, in
To find the VPA effects on non-motor PD symptoms which might be associated with mesocorticolimbic pathway, we checked the time spent in center in open field test and cognition behavior in elevated plus maze (Fig. 5C). In the open field test, LRRK2 R1441G mutant mice showed a significantly increased time spent in center compared to normal group mice (Fig. 5C). LRRK2 R1441G mutant mice stayed significantly longer time in the center area rather than the boundary area, indicating alteration of anxiety-like behavior. Interestingly, VPA significantly decreased the time spent in the center of LRRK2 R1441G mutant mice compared to vehicle group. In elevated plus maze, VPA decreased the time spent in open arms of LRRK2 R1441G mutant mice compared to vehicle group (Fig. 5C). This result indicates that VPA administration made LRRK2 R1441G mice to avoid staying in the open arms, suggesting improvement of anxiety-like behavior. Taken together, these data suggest that administration of VPA induced the improvement of motor and non-motor behavioral deficits in LRRK2 R1441G mutant mice.
DISCUSSION
Many recent studies have demonstrated neuroprotective effect of VPA in animal model of neurological diseases. Previously, we found that VPA, an anti-epileptic agent, improves impaired memory, reduces pro-inflammatory markers, and increases the expression of nerve growth factor (NGF) protein in hippocampus of Tg6799 Alzheimer’s disease (AD) model mice [37]. In this study, we addressed to determine the effects of VPA in PD model mice for the neuroprotective effects on dopaminergic neuron survival, neuroinflammation, and PD-like behaviors.
LRRK2 is the most common genetic factor associated with onset of PD. The LRRK2 is comprised of several conserved domains; a leucine-rich repeat (LRR), a Ras of complex (ROC) that belongs to the Rab GTPase superfamily, a C-terminal of Roc (COR), kinase (MAPKKK) and WD40 domain [7]. LRRK2 R1441G point mutation in ROC domain have been found as linked to both familial and sporadic forms of PD [38, 39]. LRRK2 R1441G mutation reduced the rate of GTP hydrolysis to stabilize the GTP-binding of LRRK2 protein to enhance the LRRK2 kinase enzyme activity [40]. LRRK2 R1441G mutation showed the elevated level of expression and secretion of pro-inflammatory marker, TNF-α, in LPS-stimulated microglia and increased neuronal death in cultured cortical neurons [10]. The LRRK2 R1441G mice showed diminished dopamine release level and axonal pathology of nigrostriatal DA projections at 10~12 months of age [26] and showed significant deficits in coordinated motor function by pole test and rota-rod test at 15–16 month of age [28, 41].
HDAC inhibition has been studied to understand the pathological mechanism of various neurodegenerative diseases including Parkinson’s disease. VPA has been widely used as a pan-HDAC inhibitor for various
In this study, we used PD model mice with LRRK2 R1441G mutation to determine the
When VPA was administered to LRRK R1441G mice in this study, VPA increased histone acetylation as shown in acetylated H3 protein levels, while the protein levels of HDACs were not altered. These data suggest that the regulation of histone acetylation was not through transcriptional control of HDACs, but through inhibition of enzyme activities. Surprisingly, VPA administration significantly increased TH positive DA neurons and reduced iba-1 positive activated microglia cells suggesting neuroprotective effects through microglia associated neuroinflammation. To further understand the neuroinflammatory responses after VPA administration, we performed quantitative RT-PCR for various inflammation markers. We found that mRNA levels of IL-1β, TLR2, TLR4, CD45, CD11b, TNF-α, and IκB-α were up-regulated by LRRK2 R1441G mutation. In addition, we found that VPA decreased the expression of inflammation markers including IL-1β and TLR2. Various neuroinflammatory responses have been studied to explain pathophysiological mechanism of neuroprotection against the neurodegeneration. For example, TLR2-mediated primary microglial activation has been reported to be associated with necrotic neurons [43]. TLR2 expression level was increased in the striatum of advanced PD patients [44] and in the substantia nigra and the hippocampus in the early stage of PD patients [45]. IL-1β, which binds to IL-1 receptor type 1 (IL-1R1), promotes the inflammatory response through activation of NF-κB in blood cells [46, 47]. NF-κB can enter the nucleus to upregulate genes involved T-cell development, maturation, and proliferation [46, 47]. NF-κB is also involved in diverse function in nervous system including synaptic plasticity and synaptic transmission [48, 49]. IκB-α is a key regulator of NF-κB pathway and has been used as one of the markers of NF-κB related inflammation [50]. The effect of HDAC inhibition has been studied showing repressed the inflammatory gene expression including IL-1β, TNF-α, IL-6, or interferon γ in T cell and B cell [51–53]. Seet et al. showed anti-inflammatory effects of VPA in post-operative conjunctiva [54]. Chen et al., showed that VPA reduced neuroinflammation and neuronal death in a rat chronic constriction injury model [55]. In addition, previous study found that VPA inhibited voltage-gated sodium channels blocking NMDA hypo-function related neurotoxicity [56], suggesting potential mechanism for prevention of neurodegeneration. It is also possible that altered cytokine levels affect ion channel activities and change neural activity to prevent neural toxicity and so improve motor behavior of mice.
In this study, we determined that VPA decreased mRNA expression levels of several pro-inflammatory markers including IL-1β and TLR2 in LRRK2 R1441G mice indicating reduced neuroinflammation responses which potentially provide neuroprotective environment to DA neurons. Additionally, we have observed that VPA significantly improve both motor behaviors of LRRK2 R1441G mice as shown as latency to fall in rota-rod test, rearing and wall rearing counts in open field test and non-motor behaviors including anxiety as shown as time spent in center in open field test and time spent in open arms in elevated plus maze test. These data are probably by combinatorial effects of neuroprotection, reduced neurodegeneration, and restoration of neural function with neuroregeneration. Morphological changes of neuronal/ glial cells and cellular mechanism under HDAC inhibition can be addressed for future therapeutics of PD. In summary, HDAC inhibition by systemic administration of VPA protects neurons through modulation of neuroinflammation and improves PD-like behaviors in LRRK2 R1441G mutant mice.
CONFLICT OF INTEREST
The authors declare that there are no conflicts of interest in this work.
ACKNOWLEDGEMENTS
This research was supported by grants from the National Research Foundation of Korea (NRF) funded by the Korean government (MSIP) (2017R1A2B4008456).
Figures
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Tables
Table 1 Primer sequences for RT-PCR
| Gene | Primer sequence | |
|---|---|---|
| IL-1β (Interleukin-1 beta) | Forward | 5′-CTG GTG TGT GAC GTT CCC ATT A-3′ |
| Reverse | 5′-CCG ACA GCA CGA GGC TTT-3′ | |
| TLR2 (Toll-like receptor 2) | Forward | 5′-TGC TTT CCT GCT GGA GAT TT-3′ |
| Reverse | 5′-TGT AAC GCA ACA GCT TCA GG-3′ | |
| TLR4 (Toll-like receptor 4) | Forward | 5′-ACC TGG CTG GTT TAC ACG TC-3′ |
| Reverse | 5′-CTG CCA GAG ACA TTG CAG AA-3′ | |
| SRA (Scavenger receptor A) | Forward | 5′-GAC GCT TCC AGA ATT TCA GC-3′ |
| Reverse | 5′-ATG TCC TCC TGT TGC TTT GC-3′ | |
| TNF-α (Tumor necrosis factor-α) | Forward | 5′-TTC TGT CTA CTG AAC TTC GGG-3′ |
| Reverse | 5′-GTA TGA GAT AGC AAA TCG GCT-3′ | |
| CD45 (Protein tyrosine phosphatase, receptor type, C) | Forward | 5′-CAG AGC ATT CCA CGG GTA TT-3′ |
| Reverse | 5′-GGA CCC TGC ATC TCC ATT TA-3′ | |
| CD11b (Integrin alpha M) | Forward | 5′-CAG ATC AAC AAT GTG ACC GTA TGG G-3′ |
| Reverse | 5′-CAT CAT GTC CTT GTA CTG CCG CTT G-3′ | |
| IκB-α (Nuclear factor kappa light polypeptide gene enhancer in B-cells inhibitor-α) | Forward | 5′-GAA GCC GCT GAC CAT GGA A-3′ |
| Reverse | 5′-GAT CAC AGC CAA GTG GAG TGG A-3′ | |
| GAPDH (Glyceraldehyde 3-phosphate dehydrogenase) | Forward | 5′-ATG AAT ACG GCT ACA GCA-3′ |
| Reverse | 5′-GCC CCT CCT GTT ATT ATG G-3′ |
References
- Spillantini MG, Schmidt ML, Lee VM, Trojanowski JQ, Jakes R, Goedert M (1997) Alpha-synuclein in Lewy bodies. Nature 388 : 839-840.


- Baba M, Nakajo S, Tu PH, Tomita T, Nakaya K, Lee VM, Trojanowski JQ, Iwatsubo T (1998) Aggregation of alphasynuclein in Lewy bodies of sporadic Parkinson's disease and dementia with Lewy bodies. Am J Pathol 152 : 879-884.

- Polymeropoulos MH, Lavedan C, Leroy E, Ide SE, Dehejia A, Dutra A, Pike B, Root H, Rubenstein J, Boyer R, Stenroos ES, Chandrasekharappa S, Athanassiadou A, Papapetropoulos T, Johnson WG, Lazzarini AM, Duvoisin RC, Di Iorio G, Golbe LI, Nussbaum RL (1997) Mutation in the alpha-synuclein gene identified in families with Parkinson's disease. Science 276 : 2045-2047.
- Kitada T, Asakawa S, Hattori N, Matsumine H, Yamamura Y, Minoshima S, Yokochi M, Mizuno Y, Shimizu N (1998) Mutations in the parkin gene cause autosomal recessive juvenile parkinsonism. Nature 392 : 605-608.
- Lockhart PJ, Lincoln S, Hulihan M, Kachergus J, Wilkes K, Bisceglio G, Mash DC, Farrer MJ (2004) DJ-1 mutations are a rare cause of recessively inherited early onset parkinsonism mediated by loss of protein function. J Med Genet 41 : e22.
- Valente EM, Abou-Sleiman PM, Caputo V, Muqit MM, Harvey K, Gispert S, Ali Z, Del Turco D, Bentivoglio AR, Healy DG, Albanese A, Nussbaum R, González-Maldonado R, Deller T, Salvi S, Cortelli P, Gilks WP, Latchman DS, Harvey RJ, Dallapiccola B, Auburger G, Wood NW (2004) Hereditary early-onset Parkinson's disease caused by mutations in PINK1 . Science 304 : 1158-1160.
- Zimprich A, Biskup S, Leitner P, Lichtner P, Farrer M, Lincoln S, Kachergus J, Hulihan M, Uitti RJ, Calne DB, Stoessl AJ, Pfeiffer RF, Patenge N, Carbajal IC, Vieregge P, Asmus F, Müller- Myhsok B, Dickson DW, Meitinger T, Strom TM, Wszolek ZK, Gasser T (2004) Mutations in LRRK2 cause autosomaldominant parkinsonism with pleomorphic pathology. Neuron 44 : 601-607.
- Dzamko N, Inesta-Vaquera F, Zhang J, Xie C, Cai H, Arthur S, Tan L, Choi H, Gray N, Cohen P, Pedrioli P, Clark K, Alessi DR (2012) The IkappaB kinase family phosphorylates the Parkinson's disease kinase LRRK2 at Ser935 and Ser910 during Toll-like receptor signaling. PLoS One 7 : e39132.
- Hakimi M, Selvanantham T, Swinton E, Padmore RF, Tong Y, Kabbach G, Venderova K, Girardin SE, Bulman DE, Scherzer CR, LaVoie MJ, Gris D, Park DS, Angel JB, Shen J, Philpott DJ, Schlossmacher MG (2011) Parkinson's disease-linked LRRK2 is expressed in circulating and tissue immune cells and upregulated following recognition of microbial structures. J Neural Transm (Vienna) 118 : 795-808.
- Gillardon F, Schmid R, Draheim H (2012) Parkinson's disease- linked leucine-rich repeat kinase 2(R1441G) mutation increases proinflammatory cytokine release from activated primary microglial cells and resultant neurotoxicity. Neuroscience 208 : 41-48.
- Moehle MS, Webber PJ, Tse T, Sukar N, Standaert DG, DeSilva TM, Cowell RM, West AB (2012) LRRK2 inhibition attenuates microglial inflammatory responses. J Neurosci 32 : 1602-1611.
- Brownell JE, Allis CD (1995) An activity gel assay detects a single, catalytically active histone acetyltransferase subunit in Tetrahymena macronuclei. Proc Natl Acad Sci U S A 92 : 6364-6368.
- Taunton J, Hassig CA, Schreiber SL (1996) A mammalian histone deacetylase related to the yeast transcriptional regulator Rpd3p. Science 272 : 408-411.
- Hsieh J, Nakashima K, Kuwabara T, Mejia E, Gage FH (2004) Histone deacetylase inhibition-mediated neuronal differentiation of multipotent adult neural progenitor cells. Proc Natl Acad Sci U S A 101 : 16659-16664.
- Konsoula Z, Barile FA (2012) Epigenetic histone acetylation and deacetylation mechanisms in experimental models of neurodegenerative disorders. J Pharmacol Toxicol Methods 66 : 215-220.
- Zou JY, Crews FT (2014) Release of neuronal HMGB1 by ethanol through decreased HDAC activity activates brain neuroimmune signaling. PLoS One 9 : e87915.
- Wang G, Shi Y, Jiang X, Leak RK, Hu X, Wu Y, Pu H, Li WW, Tang B, Wang Y, Gao Y, Zheng P, Bennett MV, Chen J (2015) HDAC inhibition prevents white matter injury by modulating microglia/macrophage polarization through the GSK3β/PTEN/Akt axis. Proc Natl Acad Sci U S A 112 : 2853-2858.
- Bai Y, Du S, Li F, Huang F, Deng R, Zhou J, Chen D (2017) Histone deacetylase-high mobility group box-1 pathway targeted by hypaconitine suppresses the apoptosis of endothelial cells. Exp Biol Med (Maywood) 242 : 527-535.
- Suda S, Ueda M, Nito C, Nishiyama Y, Okubo S, Abe A, Aoki J, Suzuki K, Sakamoto Y, Kimura K (2015) Valproic acid ameliorates ischemic brain injury in hyperglycemic rats with permanent middle cerebral occlusion. Brain Res 1606 : 1-8.
- Chu T, Zhou H, Lu L, Kong X, Wang T, Pan B, Feng S (2015) Valproic acid-mediated neuroprotection and neurogenesis after spinal cord injury: from mechanism to clinical potential. Regen Med 10 : 193-209.
- Chen X, Wang H, Zhou M, Li X, Fang Z, Gao H, Li Y, Hu W (2018) Valproic acid attenuates traumatic brain injuryinduced inflammation in vivo : involvement of autophagy and the Nrf2/ARE signaling pathway. Front Mol Neurosci 11 : 117.
- Chen PS, Peng GS, Li G, Yang S, Wu X, Wang CC, Wilson B, Lu RB, Gean PW, Chuang DM, Hong JS (2006) Valproate protects dopaminergic neurons in midbrain neuron/glia cultures by stimulating the release of neurotrophic factors from astrocytes. Mol Psychiatry 11 : 1116-1125.
- Wu X, Chen PS, Dallas S, Wilson B, Block ML, Wang CC, Kinyamu H, Lu N, Gao X, Leng Y, Chuang DM, Zhang W, Lu RB, Hong JS (2008) Histone deacetylase inhibitors upregulate astrocyte GDNF and BDNF gene transcription and protect dopaminergic neurons. Int J Neuropsychopharmacol 11 : 1123-1134.
- Peng GS, Li G, Tzeng NS, Chen PS, Chuang DM, Hsu YD, Yang S, Hong JS (2005) Valproate pretreatment protects dopaminergic neurons from LPS-induced neurotoxicity in rat primary midbrain cultures: role of microglia. Brain Res Mol Brain Res 134 : 162-169.
- Kidd SK, Schneider JS (2011) Protective effects of valproic acid on the nigrostriatal dopamine system in a 1-methyl- 4-phenyl-1,2,3,6-tetrahydropyridine mouse model of Parkinson's disease. Neuroscience 194 : 189-194.
- Li Y, Liu W, Oo TF, Wang L, Tang Y, Jackson-Lewis V, Zhou C, Geghman K, Bogdanov M, Przedborski S, Beal MF, Burke RE, Li C (2009) Mutant LRRK2(R1441G) BAC transgenic mice recapitulate cardinal features of Parkinson's disease. Nat Neurosci 12 : 826-828.
- Bichler Z, Lim HC, Zeng L, Tan EK (2013) Non-motor and motor features in LRRK2 transgenic mice. PLoS One 8 : e70249.
- Dranka BP, Gifford A, McAllister D, Zielonka J, Joseph J, O'Hara CL, Stucky CL, Kanthasamy AG, Kalyanaraman B (2014) A novel mitochondrially-targeted apocynin derivative prevents hyposmia and loss of motor function in the leucinerich repeat kinase 2 (LRRK2R1441G) transgenic mouse model of Parkinson's disease. Neurosci Lett 583 : 159-164.
- Castro AA, Ghisoni K, Latini A, Quevedo J, Tasca CI, Prediger RD (2012) Lithium and valproate prevent olfactory discrimination and short-term memory impairments in the intranasal 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) rat model of Parkinson's disease. Behav Brain Res 229 : 208- 215.
- Seo H, Kim W, Isacson O (2008) Compensatory changes in the ubiquitin-proteasome system, brain-derived neurotrophic factor and mitochondrial complex II/III in YAC72 and R6/2 transgenic mice partially model Huntington's disease patients. Hum Mol Genet 17 : 3144-3153.
- Kim J, Jeong YH, Lee EJ, Park JS, Seo H, Kim HS (2017) Suppression of neuroinflammation by matrix metalloproteinase- 8 inhibitor in aged normal and LRRK2 G2019S Parkinson's disease model mice challenged with lipopolysaccharide. Biochem Biophys Res Commun 493 : 879-886.
- Walf AA, Frye CA (2007) The use of the elevated plus maze as an assay of anxiety-related behavior in rodents. Nat Protoc 2 : 322-328.
- Noh H, Park C, Park S, Lee YS, Cho SY, Seo H (2014) Prediction of miRNA-mRNA associations in Alzheimer's disease mice using network topology. BMC Genomics 15 : 644.
- Nair-Roberts RG, Chatelain-Badie SD, Benson E, White-Cooper H, Bolam JP, Ungless MA (2008) Stereological estimates of dopaminergic, GABAergic and glutamatergic neurons in the ventral tegmental area, substantia nigra and retrorubral field in the rat. Neuroscience 152 : 1024-1031.
- Franklin KB, Paxinos G (2008) The mouse brain in stereotaxic coordinates. 3rd ed. Academic Press, San Diego, CA.
- Karnavas T, Pintonello L, Agresti A, Bianchi ME (2014) Histone content increases in differentiating embryonic stem cells. Front Physiol 5 : 330.
- Noh H, Seo H (2014) Age-dependent effects of valproic acid in Alzheimer's disease (AD) mice are associated with nerve growth factor (NGF) regulation. Neuroscience 266 : 255-265.
- Somme JH, Molano Salazar A, Gonzalez A, Tijero B, Berganzo K, Lezcano E, Fernandez Martinez M, Zarranz JJ, Gómez- Esteban JC (2015) Cognitive and behavioral symptoms in Parkinson's disease patients with the G2019S and R1441G mutations of the LRRK2 gene. Parkinsonism Relat Disord 21 : 494-499.
- Li X, Tan YC, Poulose S, Olanow CW, Huang XY, Yue Z (2007) Leucine-rich repeat kinase 2 (LRRK2)/PARK8 possesses GTPase activity that is altered in familial Parkinson's disease R1441C/G mutants. J Neurochem 103 : 238-247.
- Guo L, Gandhi PN, Wang W, Petersen RB, Wilson-Delfosse AL, Chen SG (2007) The Parkinson's disease-associated protein, leucine-rich repeat kinase 2 (LRRK2), is an authentic GTPase that stimulates kinase activity. Exp Cell Res 313 : 3658-3670.
- Dranka BP, Gifford A, Ghosh A, Zielonka J, Joseph J, Kanthasamy AG, Kalyanaraman B (2013) Diapocynin prevents early Parkinson's disease symptoms in the leucine-rich repeat kinase 2 (LRRK2R1441G) transgenic mouse. Neurosci Lett 549 : 57-62.
- Schwab AJ, Sison SL, Meade MR, Broniowska KA, Corbett JA, Ebert AD (2017) Decreased sirtuin deacetylase activity in LRRK2 G2019S iPSC-derived dopaminergic neurons. Stem Cell Reports 9 : 1839-1852.
- Hayward JH, Lee SJ (2014) A decade of research on TLR2 discovering its pivotal role in glial activation and neuroinflammation in neurodegenerative diseases. Exp Neurobiol 23 : 138-147.
- Drouin-Ouellet J, St-Amour I, Saint-Pierre M, Lamontagne- Proulx J, Kriz J, Barker RA, Cicchetti F (2014) Toll-like receptor expression in the blood and brain of patients and a mouse model of Parkinson's disease. Int J Neuropsychopharmacol 18 : pyu103.
- Doorn KJ, Moors T, Drukarch B, van de Berg WD, Lucassen PJ, van Dam AM (2014) Microglial phenotypes and tolllike receptor 2 in the substantia nigra and hippocampus of incidental Lewy body disease cases and Parkinson's disease patients. Acta Neuropathol Commun 2 : 90.
- Wilson KP, Black JA, Thomson JA, Kim EE, Griffith JP, Navia MA, Murcko MA, Chambers SP, Aldape RA, Raybuck SA, Livingston DJ (1994) Structure and mechanism of interleukin-1β converting enzyme. Nature 370 : 270-275.
- Thornberry NA, Bull HG, Calaycay JR, Chapman KT, Howard AD, Kostura MJ, Miller DK, Molineaux SM, Weidner JR, Aunins J, Elliston KO, Ayala JM, Casano FJ, Chin J, Ding GJ, Egger LA, Gaffney EP, Limjuco G, Palyha OC, Raju SM, Rolando AM, Salley JP, Yamin TT, Lee TD, Shively JE, Mac- Cross M, Mumford RA, Schmidt JA, Tocci MJ (1992) A novel heterodimeric cysteine protease is required for interleukin-1 beta processing in monocytes. Nature 356 : 768-774.
- Albensi BC, Mattson MP (2000) Evidence for the involvement of TNF and NF-κB in hippocampal synaptic plasticity. Synapse 35 : 151-159.
- Mattson MP, Meffert MK (2006) Roles for NF-κB in nerve cell survival, plasticity, and disease. Cell Death Differ 13 : 852-860.
- Yamamoto Y, Gaynor RB (2004) IκB kinases: key regulators of the NF-κB pathway. Trends Biochem Sci 29 : 72-79.
- Zhang Z, Zhang ZY, Fauser U, Schluesener HJ (2008) Valproic acid attenuates inflammation in experimental autoimmune neuritis. Cell Mol Life Sci 65 : 4055-4065.
- Wang X, Song Y, Jacobi JL, Tuan RS (2009) Inhibition of histone deacetylases antagonized FGF2 and IL-1β effects on MMP expression in human articular chondrocytes. Growth Factors 27 : 40-49.
- Suh HS, Choi S, Khattar P, Choi N, Lee SC (2010) Histone deacetylase inhibitors suppress the expression of inflammatory and innate immune response genes in human microglia and astrocytes. J Neuroimmune Pharmacol 5 : 521-532.
- Seet LF, Toh LZ, Finger SN, Chu SW, Wong TT (2019) Valproic acid exerts specific cellular and molecular anti-inflammatory effects in post-operative conjunctiva. J Mol Med (Berl) 97 : 63-75.
- Chen JY, Chu LW, Cheng KI, Hsieh SL, Juan YS, Wu BN (2018) Valproate reduces neuroinflammation and neuronal death in a rat chronic constriction injury model. Sci Rep 8 : 16457.
- Farber NB, Jiang XP, Heinkel C, Nemmers B (2002) Antiepileptic drugs and agents that inhibit voltage-gated sodium channels prevent NMDA antagonist neurotoxicity. Mol Psychiatry 7 : 726-733.