Articles

Article Tools
 View Full Text View Full Text |
 Abstract Abstract |
 Article as PDF Article as PDF |
 Print this Article Print this Article |
 Pubmed Pubmed |
 PMC PMC |
 PubReader PubReader |
 Export to Citation Export to Citation |
 Email Alerts Email Alerts |
 Open Access Open Access |
Share this article on :



Stats or Metrics
Article
Original Article
Exp Neurobiol 2021; 30(4): 263-274
Published online August 31, 2021
https://doi.org/10.5607/en21008
© The Korean Society for Brain and Neural Sciences
Altered Gene Expression Profiles in Neural Stem Cells Derived from Duchenne Muscular Dystrophy Patients with Intellectual Disability
Jahong Koo1,2, Subin Park1,3, Soo-Eun Sung1, Jeehun Lee4, Dae Soo Kim2,5, Jungwoon Lee2,5, Jae-Ran Lee1, Nam-Soon Kim1,2 and Da Yong Lee1,2*
1Rare Disease Research Center, Korea Research Institute of Bioscience and Biotechnology (KRIBB), Daejeon 34141, 2Department of Functional Genomics, KRIBB School of Bioscience, Korea University of Science and Technology, Daejeon 34141, 3Department of Biochemistry, Department of Medical Science, Chungnam National University School of Medicine, Daejeon 35015, 4Department of Pediatrics, Samsung Medical Center, Sungkyunkwan University School of Medicine, Seoul 06351, 5Environmental Diseases Research Center, Korea Research Institute of Bioscience and Biotechnology (KRIBB), Daejeon 34141, Korea
Correspondence to: *To whom correspondence should be addressed.
TEL: 82-42-860-4475, FAX: 82-42-879-8495
e-mail: daylee@kribb.re.kr
This is an Open Access article distributed under the terms of the Creative Commons Attribution Non-Commercial License (http://creativecommons.org/licenses/by-nc/4.0) which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
Abstract
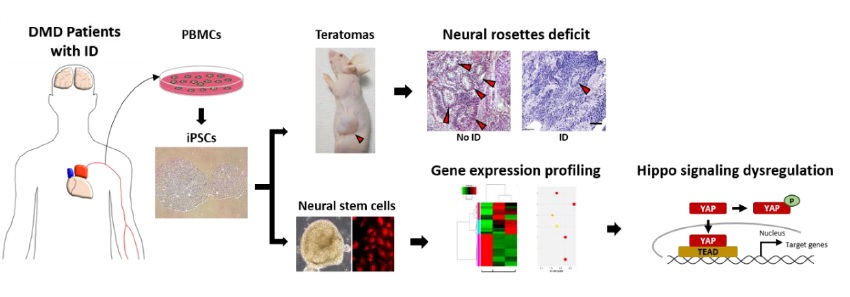
Intellectual disability (ID) is a neurodevelopmental disorder defined by below-average intelligence (intelligence quotient of <70) accompanied by adaptive behavior deficits. Defects in the functions of neural stem cells during brain development are closely linked to the pathogenesis of ID. To understand the molecular etiology of ID, we examined neural stem cells from individuals with Duchenne muscular dystrophy (DMD), a genetic disorder in which approximately one-third of the patients exhibit ID. In this study, we generated induced pluripotent stem cells from peripheral blood mononuclear cells from a normal individual and DMD patients with and without ID to identify ID-specific functional and molecular abnormalities. We found defects in neural ectoderm formation in the group of DMD patients with ID. Our transcriptome analysis of patient-derived neural stem cells revealed altered expression of genes related to the hippo signaling pathway and neuroactive ligand-receptor interaction, implicating these in the pathogenesis of ID in patients with DMD.
Graphical Abstract
Keywords: Intellectual disability, Duchenne muscular dystrophy, Induced pluripotent stem cell, Gene expression
INTRODUCTION
Intellectual disability (ID) is characterized by significant impairments in both intellectual functioning, such as reasoning, learning, and problem solving, and adaptive functioning in the activities of daily living, including social and practical skills. As the onset of ID is during childhood (before the age of 18 years), it is characterized as a developmental disorder [1, 2]. Although the incidence rate of ID continues to increase in children [3], the detailed mechanisms underlying the pathogenesis are still not fully understood. Recent evidence shows that the onset of ID and other neuropsychiatric diseases corresponds to neurodevelopmental defects caused by alteration of the physiological functions of neural stem cells (NSCs) [4, 5]. For example, altered expression of the genes associated with neurogenesis, myelination, and neural lineage differentiation has been noted in genetic diseases accompanied by ID, such as Down syndrome, fragile X-syndrome, and other X-linked psychiatric disorders [6-9].
The X-linked neuromuscular disorder Duchenne muscular dystrophy (DMD) is a severe type of muscular dystrophy that primarily affects males [10]. Males with DMD show higher incidence of ID (26~30%) and other neuropsychiatric diseases, such as autistic spectrum disorder, hyperactivity, and attention deficits (21~44%), compared with incidence rates in healthy children in the general population [11, 12]. ID and other neuropsychiatric syndromes in children with DMD result from the suppressed expression of dystrophin protein variants such as Dp71 in the brain and Dp140, the variant predominantly expressed in the central nervous system (CNS) during fetal development [12, 13]. Mice lacking full-length dystrophin (Dp427) and its variants, including Dp71, exhibit abnormal synaptic structures and synaptic plasticity in the hippocampus and the prefrontal cortex [14-16].
In this study, we generated induced pluripotent stem cells (iPSCs) using peripheral blood mononuclear cells (PBMCs) from a normal individual and from children with DMD with or without ID. The NSCs from these were analyzed to investigate the molecular signatures of ID-associated brain abnormalities during development. Specifically, we compared the patterns of gene expression in NSCs derived from patients with DMD (group with ID versus the group without ID) by transcriptome analysis.
MATERIALS AND METHODS
Human PBMC samples
A normal individual and DMD patients (with and without ID) were recruited with proper approval of Institutional Review Board (IRB) for a series of medical actions. Peripheral blood mononuclear cells (PBMCs) derived from the individuals were received from Samsung Medical Center (Seoul, Republic of Korea).
iPSC formation
iPSCs were generated from PBMCs derived from normal individual and DMD patients according to a previous report [17]. Briefly, cultured PBMCs were reprogrammed with a CytoTune–iPS 2.0 Sendai reprogramming kit (Invitrogen), which contains Yamanaka factors (Oct4, Sox2, Klf-4, and c-Myc). For reprogramming, 2.5×105 PBMCs were infected with each virus at a multiplicity of infection of 10. iPSC colonies formed from the infected PBMCs were isolated and maintained for subsequent characterization. Karyotyping analysis of iPSCs was performed by Gendix, Inc. (Seoul, Republic of Korea) using the GTG banding method.
Immunofluorescent staining
Cells cultured in well plates were fixed with 4% paraformaldehyde solution (PFA) at room temperature for 20 min and stained as described previously [18]. Primary antibodies used for staining included anti-NANOG (Invitrogen), anti-OCT4 (Santa Cruz Biotechnology), anti-TRA1-60 (Merck Millipore), anti-TRA1-81 (Merck Millipore), anti-SSEA3 (R&D systems), anti-SSEA4 (R&D systems), anti-NESTIN (abcam), anti-SOX2 (R&D systems,), anti-GATA4 (Santa Cruz Biotechnology), and anti-SMA (Sigma-Aldrich) antibodies. Signals were visualized by labeling with fluorescence conjugated appropriate secondary antibodies: Alexa FluorTM 568-conjugated goat anti-mouse and anti-rabbit IgG (H+L) and Alexa FluorTM 488-conjugated goat anti-mouse and anti-rat IgG (H+L) (Invitrogen).
Teratoma formation and analysis
iPSC colonies in 60-mm dishes were harvested by the treatment with collagenase IV (1 mg/ml; Gibco) and dispase (1 mg/ml; Gibco). Harvested colonies were washed with ES basal medium: DMEM/F12 (Gibco) containing nonessential amino acids (1%; Gibco), penicillin-streptomycin (1%; Gibco), GlutaMAX (1%; Gibco), and 2-mercaptoethanol (0.1%; Gibco). Then, colonies were resuspended with Matrigel HC (Corning) mixed with same volume of ES basal medium. Colonies were injected subcutaneously into the sides of 6-week-old BALB/c nude mice (DBL, Republic of Korea) [19]. At 6 to 12 weeks after injection, formed teratomas were collected from the injected sites and fixed with 4% PFA. Fixed teratomas were dehydrated in 70% ethanol and processed for the generation of paraffin blocks. Each block was sectioned at 5 μm, and the slides were stained with hematoxylin and eosin for further image analyses.
In vitro differentiation of iPSCs
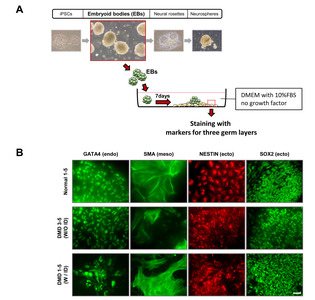
iPSC colonies were detached by treatment with collagenase IV (1 mg/ml) and dispase (1 mg/ml). Detached colonies were resuspended and maintained with differentiation medium (DMEM; Gibco) supplemented with fetal bovine serum (10%; Gibco) and penicillin-streptomycin (1%; Gibco) in low-attachment dishes. After 7 days of culture in suspension, formed embryoid body (EB) spheres were collected and plated on gelatin-coated (2%; Sigma-Aldrich) plates. After 7 days, the attached cells were fixed with 4% PFA for further analyses [20, 21].
EB and NSC formation
Human EBs and NSCs were formed as previously described [22]. Briefly, iPSCs were detached and cultured in suspension for 6 days. To form EBs, detached iPSC colonies were cultured in suspension with DMEM/F12 medium supplemented with knockout serum replacement (10%; Gibco) and penicillin-streptomycin (1%). EB spheres were attached to Matrigel (1%; Corning) -coated culture plates and maintained with NSC medium until appropriately sized neural rosettes were formed. NSC medium was supplemented with B-27 (2%; Gibco), N-2 (1%; Gibco), basic fibroblast growth factor (bFGF; 20 μg/ml; Gibco), and penicillin-streptomycin (1%) in DMEM/F12. To form neurospheres, neural rosettes were detached mechanically and cultured in suspension with NSC medium.
RNA sequencing
For RNA sequencing (RNA-seq), RNA samples were analyzed using an Agilent 2100 Bioanalyzer system (Agilent Biotechnologies). RNA samples with high quality (RNA integrity number ≥7.5) were used for the subsequent mRNA sample preparation for sequencing. Libraries were prepared using an Illumina TruSeq library preparation kit as per the manufacturer’s specifications. RNA-seq was performed on Illumina HiSeq 2500 machines (Illumina) using the standard Illumina RNA-seq protocol with a read length of 2×100 bases [23].
Bioinformatic analysis
The quality of sequencing data was evaluated with NGS QC Toolkit v2.3.3. Adapters were removed using Cutadapt v1.18 with default settings, and low-quality sequences were also trimmed via Sickle v1.33 with a Phred quality threshold score of 20. The trimmed reads containing any ambiguous character (such as N) and less than 50 bp were excluded. After preprocessing of raw reads, the clean reads were mapped to the reference genome with HISAT2 v2.0.5 with default parameter settings, and StringTie v2.1.0 was applied using the reference annotation file to estimate the expression levels of all genes and transcripts. For the calculation of differentially expressed genes (DEGs), Cuffdiff v2.2.1 differences according to fragments per kilobase per million (FPKM) values showing at least 2-fold changes with the p values lower than 0.05 were considered significant. Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment analysis was performed using WebGestalt v0.4.4 R package [24].
Western blotting
Western blotting was performed as described previously [18] using the following antibodies: anti-YAP (Cell Signaling Technology), anti-phospho-YAP (Cell Signaling Technology), and anti-GAPDH (Cell Signaling Technology). To detect the signals, appropriate horseradish peroxidase-conjugated secondary antibodies were used and visualized with PierceTM ECL Western blotting substrate (Thermo Fisher).
qRT-PCR
Total RNA was isolated from NSCs using Trizol (Thermo Fisher). Quantitative real-time PCR (qRT-PCR) was performed with cDNA synthesized using TOPscriptTM RT DryMIX (Enzynomics) and specific primers. The reactions were performed with SYBR green master mix (Applied Biosystems) according to the manufacturer’s directions. A StepOnePlusTM real-time PCR system (Applied Biosystems) was used. All results were normalized to the value for
Statistical analysis
Results are shown as the means±standard errors of the means (SEMs). Statistical analysis was performed using one-way ANOVA with Bonferroni’s and Dunnett’s post hoc test. p values of <0.05 were considered significant.
RESULTS
iPSC generation from PBMCs from children with DMD
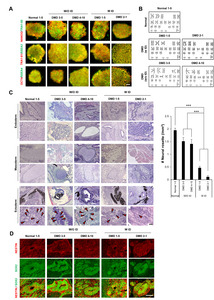
We generated iPSCs using PBMCs from children with DMD (two with ID and two without ID) and from a normal individual (Table 1). To examine whether PBMC-derived iPSC colonies formed via Yamanaka factors (
iPSCs derived from children with ID show defects in neural lineage differentiation
iPSCs derived from patients with neurological diseases often show defects in neural lineage differentiation [25]. To examine whether the differentiation potential was changed in iPSCs from DMD patients with ID, we quantified the number of neural rosettes in isolated teratomas. We found significantly fewer neural rosettes in teratomas derived from the DMD patients with ID (DMD1 and DMD2) than in those from DMD patients without ID (DMD3 and DMD4) and the normal control (Fig. 1C). To examine whether these neural rosettes were composed of NSCs, adjacent sections of teratomas were immunostained with NESTIN and SOX2 antibodies. We found clear expression of both NESTIN and SOX2 in neural rosettes (Fig. 1D). In addition, we also found defects in axon formation in neurons derived from the DMD patients with ID compared with the ones from the patients without ID (unpublished data).
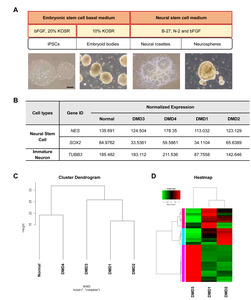
To understand the abnormalities in neural lineage differentiation at the molecular level, we performed transcriptome analysis using NSCs (neurospheres) generated from the iPSCs through the formation of EBs and neural rosettes (Fig. 3A). The levels of
Gene expression pattern was altered in NSCs derived from DMD patients with ID
To assess the dissimilarity between the DMD patients with ID and those without ID, we next performed a hierarchical clustering analysis of the transcriptome data. NSCs from the DMD patients with ID (DMD1 and DMD2) clustered separately from the sample from a DMD patient without ID (DMD3), whereas the sample from the other DMD patient without ID (DMD4) clustered with the sample from the normal individual instead of the samples from DMD patients (Fig. 3C). According to these clustering data, we used DMD1, -2, and -3 for the following analyses to identify ID-specific alteration of gene expression in the DMD disease condition. Our DEG data show that 374 genes were differentially expressed (>2- or <0.5-fold change) between the samples from DMD patients with ID (DMD1 and -2) and those from the DMD patient without ID (DMD3) (Fig. 3D). These data show an altered gene expression pattern in the NSCs derived from DMD patients with ID.
Intracellular signaling pathways are altered in NSCs from DMD patients with ID
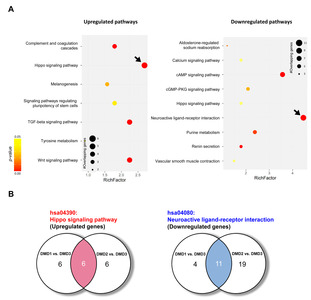
To determine the key signaling pathways responsible for ID pathogenesis in DMD, we next performed a pathway analysis using DEGs from RNA-seq data from DMD patients with and without ID. A KEGG pathway enrichment analysis revealed altered expression of genes in various signaling pathways (Fig. 4). The highest increase in expression was for hippo signaling pathway-related genes, and the greatest decrease in expression was for the genes involved in neuroactive ligand-receptor interactions (with the highest gene counts and the lowest p values; Fig. 4A and Table 3). In this analysis, we found 6 commonly upregulated hippo signaling genes and 11 commonly downregulated neuroactive ligand-receptor interaction genes in both DMD1 and DMD2 compared with expression levels in DMD3 (Fig. 4B and Table 4).
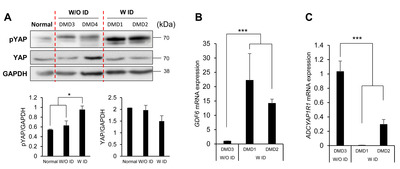
To examine whether the activity of the hippo signaling pathway was altered in NSCs from the DMD patients with ID, we next analyzed the phosphorylation level of YAP, which is a known effector molecule of this pathway. We found that the level of phospho-YAP was higher in NSCs from the DMD patients with ID than in cells from individuals without ID (Fig. 5A). An upregulation of the hippo signaling pathway was further supported by qRT-PCR, which showed a significant increase of
DISCUSSION
In this study, we sought to identify genes and signaling pathways associated with ID pathogenesis in children with DMD using patient-derived iPSCs and NSCs. Here, we demonstrate that cells derived from DMD patients with ID show reduced neural ectoderm formation and expression of the immature neuronal marker
It is important to identify the signaling pathways and related genes that are dysregulated under disease conditions as a means to elucidate the molecular mechanism(s) underlying disease onset. Our network analysis data reveal the suppression of
In accordance with our network analysis data, an alteration of genes related to neuroactive ligand-receptor interaction pathway is noted in a database of patients with Down syndrome [32]. In addition, neurons derived from the iPSCs of patients with autism spectrum disorder accompanied by ID also show altered expression of genes associated with the neuroactive ligand-receptor interaction pathway [33].
The present study reveals that the expression of genes involved in the hippo signaling pathway is increased in NSCs from individuals with ID, implicating this pathway in the pathogenesis of this developmental disorder. Especially,
ACKNOWLEDGEMENTS
This study was supported by the Korea Research Institute of Bioscience and Biotechnology (KRIBB) Research Initiative Program (KGM5222113), the Brain Research Program of the National Research Foundation (NRF) funded by the Korean government (MSIT) [grant numbers NRF-2019M3C7A1031534 (D.Y.L.), NRF-2015M3C7A1029113 (J.-R.L.)] and Basic Science Research Program of the NRF funded by Korean government (Ministry of Education) [grant number NRF-2019R1I1A2A01063642 (D.Y.L.)].
Figures
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Tables
Participants in this study
| ID | Disease | Intellectual disability | Age | Sex (M/F) |
|---|---|---|---|---|
| Normal | - | - | 46 | M |
| DMD 1 | Duchenne muscular dystrophy | Y | 4 | M |
| DMD2 | Duchenne muscular dystrophy | Y | 5 | M |
| DMD3 | Duchenne muscular dystrophy | N | 12 | M |
| DMD4 | Duchenne muscular dystrophy | N | 10 | M |
mRNA levels of neural lineage cell markers in NSCs derived from a normal individual and DMD patients with/without ID
| Cell type | Gene | Normalized expression | ||||
|---|---|---|---|---|---|---|
| Normal | DMD3 | DMD4 | DMD1 | DMD2 | ||
| Neuron | 8.04526 | 15.1402 | 10.1815 | 10.8204 | 3.89087 | |
| 32.6147 | 30.3051 | 34.0096 | 19.3397 | 35.3675 | ||
| 4.7724 | 1.89539 | 2.02749 | 1.80784 | 0.627869 | ||
| 3.76322 | 0.328347 | 1.43081 | 0.833755 | 1.58182 | ||
| 0.058355 | 0.053428 | 0 | 0 | 0 | ||
| Glia | 6.43223 | 3.96549 | 1.51845 | 1.52705 | 8.90372 | |
| 0.086698 | 0.059582 | 0.062215 | 0.100841 | 0.060418 | ||
| 1,240.69 | 1,171.03 | 861.344 | 1,382.02 | 720.448 | ||
| 0.839448 | 0.411992 | 0.947939 | 2.20389 | 0.250711 | ||
| 0.458501 | 0.395369 | 0.774859 | 0.577222 | 0.225543 | ||
List of altered signaling pathways in DMD patients with ID versus in those without ID
| Term (upregulated in ID) | Counts | p-value | Benjamini | |||||
| DMD1 vs. DMD3 | DMD2 vs. DMD3 | DMD1 vs. DMD3 | DMD2 vs. DMD3 | DMD1 vs. DMD3 | DMD2 vs. DMD3 | |||
| hsa04390: Hippo signaling pathway | 12§ | 12§ | 6.E-04 | 1.E-03 | 2.E-02 | 3.E-01 | ||
| hsa04350:TGF-beta signaling pathway | 10 | 6 | 1.E-04 | 6.E-02 | 7.E-03 | 8.E-01 | ||
| hsa04610:Complement and coagulation cascades | 8 | 5 | 9.E-04 | 1.E-01 | 3.E-02 | 8.E-01 | ||
| hsa04310:Wnt signaling pathway | 10 | 9 | 4.E-03 | 2.E-02 | 7.E-02 | 7.E-01 | ||
| hsa04916:Melanogenesis | 7 | 8 | 3.E-02 | 1.E-02 | 3.E-01 | 8.E-01 | ||
| hsa04550:Signaling pathways regulating pluripotency of stem cells | 8 | - | 3.E-02 | - | 3.E-01 | - | ||
| hsa00350:Tyrosine metabolism | 4 | 4 | 4.E-02 | 6.E-02 | 3.E-01 | 8.E-01 | ||
| Term (downregulated in ID) | Counts | p-value | Benjamini | |||||
| DMD1 vs. DMD3 | DMD2 vs. DMD3 | DMD1 vs. DMD3 | DMD2 vs. DMD3 | DMD1 vs. DMD3 | DMD2 vs. DMD3 | |||
| hsa04080: Neuroactive ligand-receptor interaction | 15§ | 30§ | 2.E-05 | 1.E-08 | 4.E-03 | 3.E-06 | ||
| hsa04924:Renin secretion | 6 | 14 | 2.E-03 | 9.E-08 | 1.E-01 | 9.E-06 | ||
| hsa00230:Purine metabolism | 8 | - | 1.E-02 | - | 3.E-01 | - | ||
| hsa04960:Aldosterone-regulated sodium reabsorption | 4 | 6 | 2.E-02 | 8.E-03 | 4.E-01 | 7.E-02 | ||
| hsa04022:cGMP-PKG signaling pathway | 7 | 12 | 3.E-02 | 2.E-02 | 4.E-01 | 1.E-01 | ||
| hsa04390:Hippo signaling pathway | 6 | - | 6.E-02 | - | 7.E-01 | - | ||
| hsa04270:Vascular smooth muscle contraction | 5 | 11 | 8.E-02 | 5.E-03 | 8.E-01 | 6.E-02 | ||
| hsa04020:Calcium signaling pathway | 6 | 15 | 1.E-01 | 2.E-03 | 8.E-01 | 2.E-02 | ||
List of commonly upregulated and downregulated genes in the DMD patients with ID compared with the group without ID
| Signaling pathway | Gene | Fold change | |
|---|---|---|---|
| DMD1 vs. DMD3 | DMD2 vs. DMD3 | ||
| Hippo signaling pathway | 6.734365 | 9.551726 | |
| 5.302346 | 22.71738 | ||
| 3.75975 | 6.783691 | ||
| 2.792583 | 4.386734 | ||
| 2.571657 | 4.092676 | ||
| 2.482384 | 2.31685 | ||
| Neuroactive ligand-receptor interaction | 0.127822 | 0.081282 | |
| 0.248777 | 0.139321 | ||
| 0.300998 | 0.157976 | ||
| 0.248864 | 0.264498 | ||
| 0.274703 | 0.245571 | ||
| 0.36375 | 0.263247 | ||
| 0.36718 | 0.319653 | ||
| 0.420563 | 0.208747 | ||
| 0.453914 | 0.269158 | ||
| 0.478967 | 0.35384 | ||
| 0.469736 | 0.478906 | ||
References
- Vasudevan P, Suri M (2017) A clinical approach to developmental delay and intellectual disability. Clin Med (Lond) 17:558-561



- American Association on Intellectual and Developmental Disabilities (AAIDD) (2010) Intellectual disability: definition, classification, and systems of supports. 11th ed. AAIDD, Washington, D.C
- Zablotsky B, Black LI, Maenner MJ, Schieve LA, Danielson ML, Bitsko RH, Blumberg SJ, Kogan MD, Boyle CA (2019) Prevalence and trends of developmental disabilities among children in the United States: 2009-2017. Pediatrics v:e20190811
- Bowers M, Liang T, Gonzalez-Bohorquez D, Zocher S, Jaeger BN, Kovacs WJ, Röhrl C, Cramb KML, Winterer J, Kruse M, Dimitrieva S, Overall RW, Wegleiter T, Najmabadi H, Semenkovich CF, Kempermann G, Földy C, Jessberger S (2020) FASN-dependent lipid metabolism links neurogenic stem/progenitor cell activity to learning and memory deficits. Cell Stem Cell 27:98-109.e11
- Qin T, Yuan Z, Yu J, Fu X, Deng X, Fu Q, Ma Z, Ma S (2020) Saikosaponin-d impedes hippocampal neurogenesis and causes cognitive deficits by inhibiting the survival of neural stem/progenitor cells via neurotrophin receptor signaling in mice. Clin Transl Med 10:e243
- Sobol M, Klar J, Laan L, Shahsavani M, Schuster J, Annerén G, Konzer A, Mi J, Bergquist J, Nordlund J, Hoeber J, Huss M, Falk A, Dahl N (2019) Transcriptome and proteome profiling of neural induced pluripotent stem cells from individuals with Down syndrome disclose dynamic dysregulations of key pathways and cellular functions. Mol Neurobiol 56:7113-7127
- Li M, Shin J, Risgaard RD, Parries MJ, Wang J, Chasman D, Liu S, Roy S, Bhattacharyya A, Zhao X (2020) Identification of FMR1-regulated molecular networks in human neurodevelopment. Genome Res 30:361-374
- Liu H, Barnes J, Pedrosa E, Herman NS, Salas F, Wang P, Zheng D, Lachman HM (2020) Transcriptome analysis of neural progenitor cells derived from Lowe syndrome induced pluripotent stem cells: identification of candidate genes for the neurodevelopmental and eye manifestations. J Neurodev Disord 12:14
- Liu B, Li Y, Stackpole EE, Novak A, Gao Y, Zhao Y, Zhao X, Richter JD (2018) Regulatory discrimination of mRNAs by FMRP controls mouse adult neural stem cell differentiation. Proc Natl Acad Sci U S A 115:E11397-E11405
- National Institute of Neurological Disorders and Stroke (2016) National Institute of Neurological Disorders and Stroke (2016) Muscular dystrophy: hope through research [Internet]. National Institute of Neurological Disorders and Stroke, Bethesda, MD.
Available from: https://web.archive.org/web/20160930165657/http://www.ninds.nih.gov/disorders/md/detail_md.htm#180483171 - Ricotti V, Mandy WP, Scoto M, Pane M, Deconinck N, Messina S, Mercuri E, Skuse DH, Muntoni F (2016) Neurodevelopmental, emotional, and behavioural problems in Duchenne muscular dystrophy in relation to underlying dystrophin gene mutations. Dev Med Child Neurol 58:77-84
- Naidoo M, Anthony K (2020) Dystrophin Dp71 and the neuropathophysiology of Duchenne muscular dystrophy. Mol Neurobiol 57:1748-1767
- Moizard MP, Billard C, Toutain A, Berret F, Marmin N, Moraine C (1998) Are Dp71 and Dp140 brain dystrophin isoforms related to cognitive impairment in Duchenne muscular dystrophy?. Am J Med Genet 80:32-41
- Miranda R, Nudel U, Laroche S, Vaillend C (2011) Altered presynaptic ultrastructure in excitatory hippocampal synapses of mice lacking dystrophins Dp427 or Dp71. Neurobiol Dis 43:134-141
- Daoud F, Candelario-Martínez A, Billard JM, Avital A, Khelfaoui M, Rozenvald Y, Guegan M, Mornet D, Jaillard D, Nudel U, Chelly J, Martínez-Rojas D, Laroche S, Yaffe D, Vaillend C (2008) Role of mental retardation-associated dystrophin-gene product Dp71 in excitatory synapse organization, synaptic plasticity and behavioral functions. PLoS One 4:e6574
- Chaussenot R, Amar M, Fossier P, Vaillend C (2019) Dp71-dystrophin deficiency alters prefrontal cortex excitation-inhibition balance and executive functions. Mol Neurobiol 56:2670-2684
- Yang W, Mills JA, Sullivan S, Liu Y, French DL, Gadue P (2008) iPSC reprogramming from human peripheral blood using sendai virus mediated gene transfer. StemBook (Stem Cell Research Community, ed). Harvard Stem Cell Institute, Cambridge, MA
- Jeong B, Kim TH, Kim DS, Shin WH, Lee JR, Kim NS, Lee DY (2019) Spastin contributes to neural development through the regulation of microtubule dynamics in the primary cilia of neural stem cells. Neuroscience 411:76-85
- Lee J, Lee J, Cho YS (2018) Peroxisome proliferator-activated receptor α agonist and its target Nanog cooperate to induce pluripotency. J Clin Med 7:488
- Jeon H, Kim JY, Choi JK, Han E, Song CL, Lee J, Cho YS (2018) Effects of the extracts from fruit and stem of
Camellia japonica on induced pluripotency and wound healing. J Clin Med 7:449
- Seol B, Kim YD, Cho YS (2021) Modeling sialidosis with neural precursor cells derived from patient-derived induced pluripotent stem cells. Int J Mol Sci 22:4386
- Kim TH, Sung SE, Cheal Yoo J, Park JY, Yi GS, Heo JY, Lee JR, Kim NS, Lee DY (2018) Copine1 regulates neural stem cell functions during brain development. Biochem Biophys Res Commun 495:168-173
- Son MY, Jung CR, Kim DS, Cho HS (2018) Comparative in silico profiling of epigenetic modifiers in human tissues. Mol Biol Rep 45:309-314
- Lee H, Son YS, Lee MO, Ryu JW, Park K, Kwon O, Jung KB, Kim K, Ryu TY, Baek A, Kim J, Jung CR, Ryu CM, Park YJ, Han TS, Kim DS, Cho HS, Son MY (2020) Low-dose interleukin-2 alleviates dextran sodium sulfate-induced colitis in mice by recovering intestinal integrity and inhibiting AKT-dependent pathways. Theranostics 10:5048-5063
- Son MY, Kwak JE, Seol B, Lee DY, Jeon H, Cho YS (2015) A novel human model of the neurodegenerative disease GM1 gangliosidosis using induced pluripotent stem cells demonstrates inflammasome activation. J Pathol 237:98-110
- Annese T, Corsi P, Ruggieri S, Tamma R, Marinaccio C, Picocci S, Errede M, Specchia G, De Luca A, Frassanito MA, Desantis V, Vacca A, Ribatti D, Nico B (2016) Isolation and characterization of neural stem cells from dystrophic mdx mouse. Exp Cell Res 343:190-207
- Romo-Yáñez J, Rodríguez-Martínez G, Aragón J, Siqueiros-Márquez L, Herrera-Salazar A, Velasco I, Montanez C (2020) Characterization of the expression of dystrophins and dystrophin-associated proteins during embryonic neural stem/progenitor cell differentiation. Neurosci Lett 736:135247
- Stagni F, Giacomini A, Emili M, Guidi S, Bartesaghi R (2018) Neurogenesis impairment: an early developmental defect in Down syndrome. Free Radic Biol Med 114:15-32
- Hagerman RJ, Berry-Kravis E, Hazlett HC, Bailey DB Jr, Moine H, Kooy RF, Tassone F, Gantois I, Sonenberg N, Mandel JL, Hagerman PJ (2017) Fragile X syndrome. Nat Rev Dis Primers 3:17065
- Adnani L, Langevin LM, Gautier E, Dixit R, Parsons K, Li S, Kaushik G, Wilkinson G, Wilson R, Childs S, Nguyen MD, Journot L, Dehay C, Schuurmans C (2015) Zac1 regulates the differentiation and migration of neocortical neurons via Pac1. J Neurosci 35:13430-13447
- Pinson A, Namba T, Huttner WB (2019) Malformations of human neocortex in development - their progenitor cell basis and experimental model systems. Front Cell Neurosci 13:305
- Chen M, Wang J, Luo Y, Huang K, Shi X, Liu Y, Li J, Lai Z, Xue S, Gao H, Chen A, Chen D (2018) Identify Down syndrome transcriptome associations using integrative analysis of microarray database and correlation-interaction network. Hum Genomics 12:2
- Liu X, Campanac E, Cheung HH, Ziats MN, Canterel-Thouennon L, Raygada M, Baxendale V, Pang AL, Yang L, Swedo S, Thurm A, Lee TL, Fung KP, Chan WY, Hoffman DA, Rennert OM (2017) Idiopathic autism: cellular and molecular phenotypes in pluripotent stem cell-derived neurons. Mol Neurobiol 54:4507-4523
- Hanel ML, Hensey C (2006) Eye and neural defects associated with loss of GDF6. BMC Dev Biol 6:43
- Masliantsev K, Karayan-Tapon L, Guichet PO (2021) Hippo signaling pathway in gliomas. Cells 10:184
- Mo JS, Park HW, Guan KL (2014) The Hippo signaling pathway in stem cell biology and cancer. EMBO Rep 15:642-656
- Pan D (2010) The hippo signaling pathway in development and cancer. Dev Cell 19:491-505
- Ouyang T, Meng W, Li M, Hong T, Zhang N (2020) Recent advances of the Hippo/YAP signaling pathway in brain development and glioma. Cell Mol Neurobiol 40:495-510
- Mueller KA, Glajch KE, Huizenga MN, Wilson RA, Granucci EJ, Dios AM, Tousley AR, Iuliano M, Weisman E, LaQuaglia MJ, DiFiglia M, Kegel-Gleason K, Vakili K, Sadri-Vakili G (2018) Hippo signaling pathway dysregulation in human Huntington's disease brain and neuronal stem cells. Sci Rep 8:11355
- Tanaka H, Homma H, Fujita K, Kondo K, Yamada S, Jin X, Waragai M, Ohtomo G, Iwata A, Tagawa K, Atsuta N, Katsuno M, Tomita N, Furukawa K, Saito Y, Saito T, Ichise A, Shibata S, Arai H, Saido T, Sudol M, Muramatsu SI, Okano H, Mufson EJ, Sobue G, Murayama S, Okazawa H (2020) YAP-dependent necrosis occurs in early stages of Alzheimer's disease and regulates mouse model pathology. Nat Commun 11:507